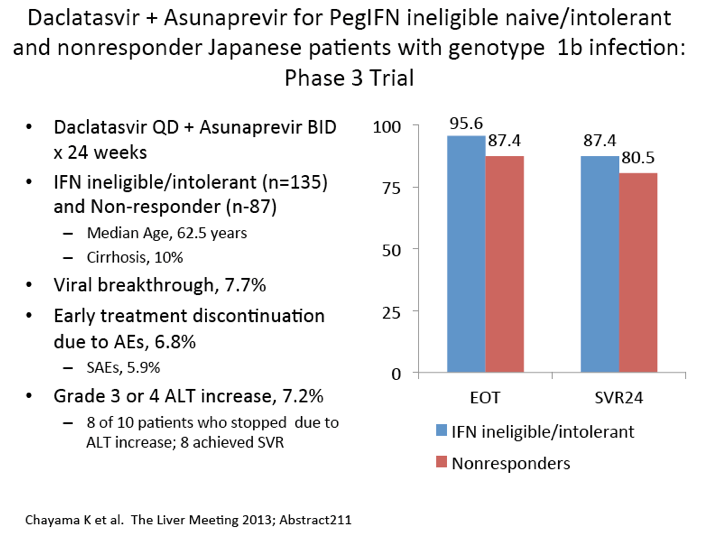
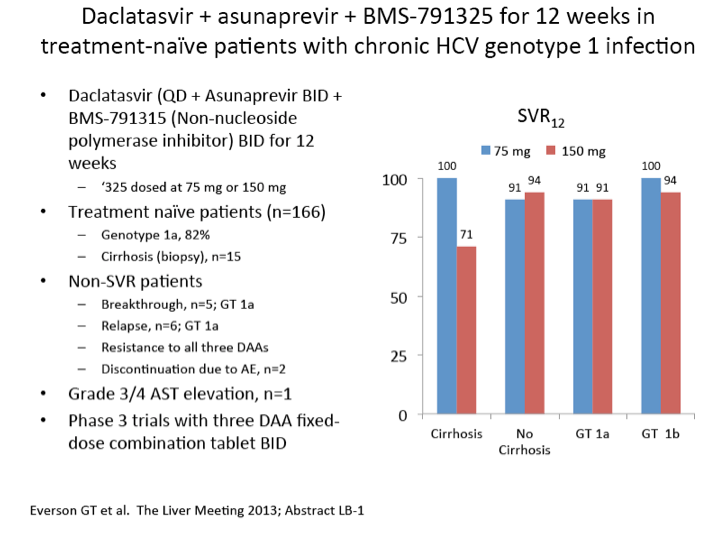
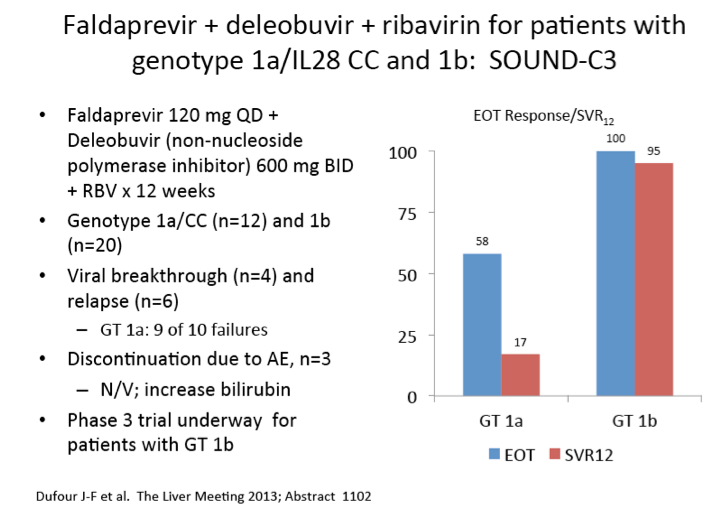
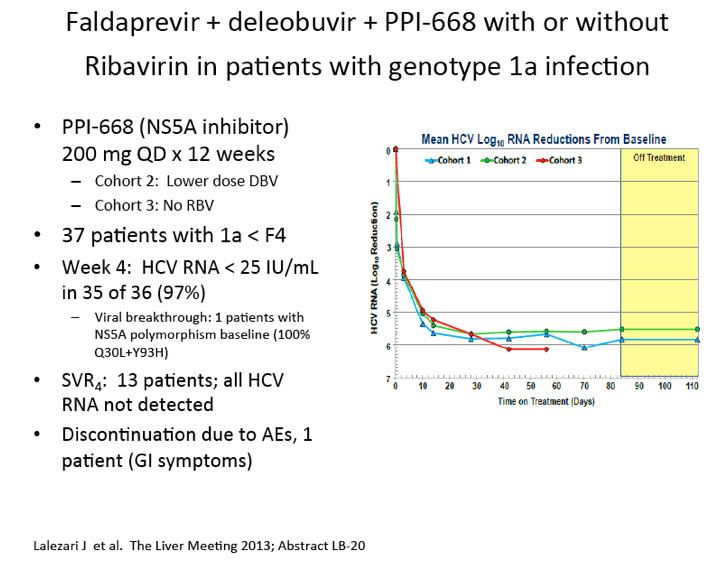
 |
|
|
| |
Hepatitis Debrief by Mark Sulkowski MD
|
| |
| |
Reported by Jules Levin
from Jules: In this very well done AASLD Hepatitis Debrief presented at the very end of the conference by Dr Mark Sulkowski he takes us through the key presentations & messages presented at this AASLD. The conference was breathtaking because of the new data from studies of new oral HCV treatments (DAAs, direct acting antivirals). This update covers new data reviews for Simeprevir & Faldaprevir in combination with Peg/Rbv, Sofosbuvir in combination with Peg/Rbv, and coinfection data including Sofosbuvir+Rbv for 24 weeks in GT 1 coinfected. As well key GT3 data are reported from the VALENCE Study with Sofosbuvir+Peg/Rbv. The SYNERGY Study from the NIH in African-Americans provided data supporting that DAA oral therapies will be equally effective for African-Americans (see the report, link below). The COSMOS Study, cohorts 1 & 2 report results of the IFN-free regimen of Simeprevir+Sofosbuvir in null responders & advanced disease with 96-100% SVR rates. Previously reported Daclatasvir+Sofosbuvir IFN-free results showed 100% SVR in HCV treatment-naives & for those previously treated & who failed telaprevir & boceprevir. Several select epidemiological studies are covered here including confirming the risk for HCC even after SVR highlighting the compelling key to avoid undue delay of HCV therapy, and the Global Burden of HCC. The FDA is expected to announce by the end of November & the beginning of December that Simeprevir+Peg/Rbv will be approved, that Sofosbuvir+Peg/Rbv & Sofosbuvir+RBv will be approved. In 2014 both Abbvie & Gilead will be submitting applications to the FDA for approvals for their IFN-free oral regimens to treat HCV for 12 weeks. In development, earlier developmental stages are BMS, Merck, & Boehringer Ingelheim with similarly promising IFN-free all oral IFN-free regimens, with all these 3 companies's regimens presented at this conference, phase 2 data, and mentioned below.
Here is a link to the NATAP conference coverage which had already reported these data during the conference in real-time:
64rd Annual Meeting of the American Association for the Study of Liver Diseases
Washington, DC Nov 1-5 2013
Presented at AASLD 2013 Nov 1-4 Wash DC by
Mark Sulkowski, MD
Professor of Medicine
Division of Infectious Diseases and Gastroenterology/Hepatology
Johns Hopkins University School of Medicine
Baltimore, MD.




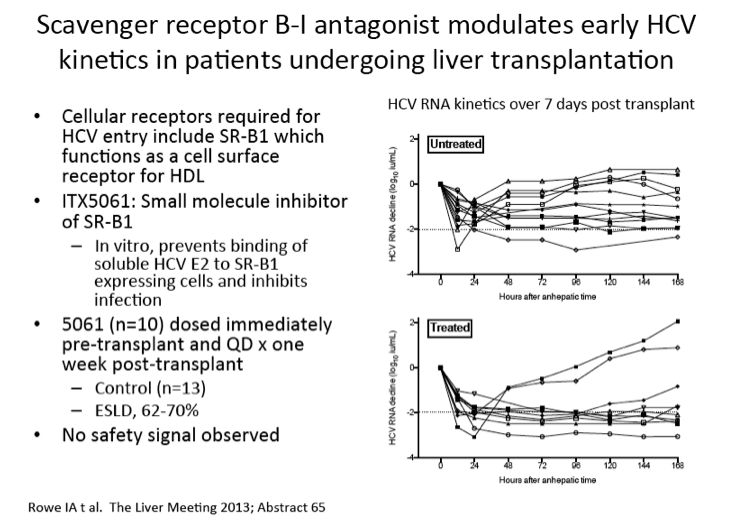
Scavenger receptor B-I antagonist ITX5061 modulates early HCV kinetics in patients undergoing liver transplantation: results of a phase Ib clinical trial
PRESENTER: Ian Rowe
AUTHOR/INSTITUTIONS: I.A. Rowe, M.J. Armstrong, R. Parker, K. Guo, D. Barton, D.H. Adams, D.J. Mutimer, Centre for Liver Research and NIHR Birmingham Liver Biomedical Research Unit, University of Birmingham, Birmingham, UNITED KINGDOM|I.A. Rowe, M.J. Armstr
SPONSORSHIP: iTherX, Inc., and NIHR Birmingham Liver Biomedical Research Unit
ABSTRACT BODY:
Prevention of recurrent HCV infection after liver transplantation (LT) is a major unmet clinical need. ITX5061 is a small molecule antagonist of scavenger receptor B-I (SR-BI) that prevents HCV entry and infection in vitro. The aim of this phase Ib study was to determine safety and efficacy of ITX5061 to prevent HCV allograft infection. Phase Ib single centre prospective open label study including 23 consecutive patients (21 males) undergoing LT. The first 13 control patients did not receive study drug. The subsequent 10 patients received ITX5061 150mg orally immediately pre-LT, post-LT and daily for 1 week. Plasma HCV RNA was quantified at 13 defined timepoints during the first week and weekly thereafter for one month, allowing detailed viral kinetic analysis. ITX5061 plasma concentrations before and after LT were measured with liquid chromatography/mass spectrometry. Clinicaltrials.gov NCT01292824. The majority of recipients were infected with HCV G1 (13/23). All patients survived 1 month after LT. ITX5061 treatment was well tolerated with no difference in the nature or frequency of adverse events between treated and untreated subjects. Oral absorption of ITX5061 was demonstrated with measurable plasma concentrations before and daily for one week after LT. HCV RNA declined during the first 24 hours but remained detectable for all patients during treatment and follow-up. Compared with values measured during the anhepatic phase of surgery, the median reduction in HCV RNA was greater for treated patients at all time-points (approximately 1log10 difference during treatment). The maximal decline in HCV RNA was greater than 2log10 in 8/10 treated patients compared to 6/13 untreated patients (Figure). In G1 patients (n=6) 7 days treatment was associated with a sustained reduction in HCV RNA (all greater than 2log10) compared with the control group (n=7), none of whom achieved a 2log10 reduction at any time. Following treatment withdrawal HCV RNA increased in all patients. Treatment with ITX5061, an inhibitor of HCV entry, is safe and well tolerated in LT. ITX5061 influences HCV RNA kinetics and a significant reduction in HCV RNA titres during treatment was observed. These findings support further investigation into the efficacy of ITX5061 in HCV infected patients undergoing LT.
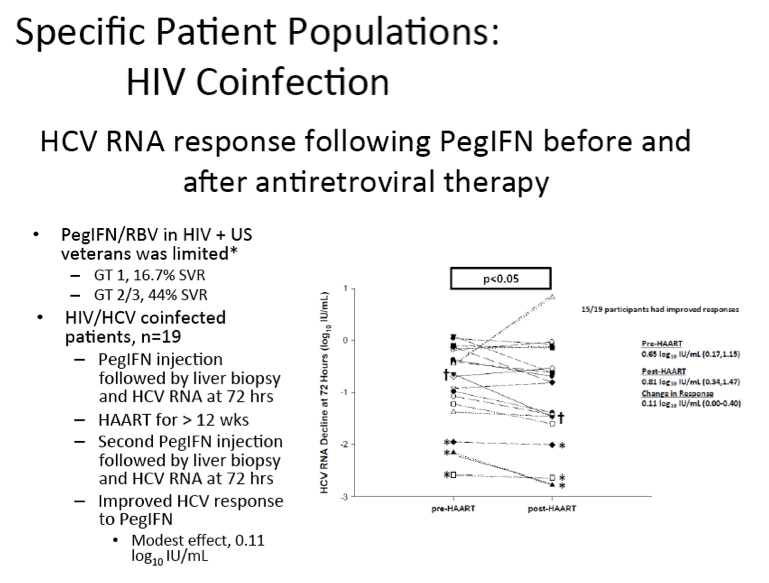
Differential Impact of Type 1 Interferon on Chronic Hepatitis C Infection in HIV Co-infection, Pre- and Post-HAART
Ashwin Balagopal1, Abraham J. Kandathil1, Johnanathan Wood1, Fuat Kurbanov1, Jeffrey Quinn1, Justin Richer1, Yvonne M. Higgins1, Lois Eldred1, Zhiping Li1, Mark S. Sulkowski1, David L. Thomas1
ABSTRACT BODY: Viral dynamic studies were performed pre- and post-
antiretroviral therapy (ART) in HIV-HCV co-infected persons to test whether HIV suppression would improve the response of HCV to interferon alfa. Persons with chronic HIV-HCV co-infection were recruited if they were treatment naïve for HCV and had no ART for HIV within 12 months AND cumulatively for <24 months, were HBsAg negative, and had CD4+ T cell counts >200/mm3. A baseline viral dynamic study (pre-ART) was performed at the Johns Hopkins Hospital Clinical Research Unit; HCV RNA was measured at baseline (time-zero) and at 12, 24, 48, and 72 hours after 1.5μg/kg of peginterferon (IFN) alfa 2b. After 14 days, ART consisting of raltegravir, tenofovir, and emtricitabine was given. When HIV RNA levels were <400 c/ml for ≥12 weeks, subjects were restudied in an identical IFN study (post-ART). Liver biopsies were performed by mini-laparoscopy 2-4 hours before each IFN dose. Of a planned 20 participants, 20 gave informed consent and were enrolled; one did not complete the study. At baseline, median (range) age was 49.1 years (21.4-60.6), 4/19 (21%) were female, and 12/19 (63.2%) were black. Median (IQR) CD4+ T cell count and HIV RNA level were 425 cells/μL (219-690) and 4.27 log10 cp/mL (2.91-5.44), respectively. Most subjects had HCV genotype 1a infection (15/19) with median (IQR) HCV RNA levels of 6.83 log10 IU/mL (6.04-7.62); 14/19 (73.7%) had Metavir scores<2, and none had cirrhosis. The post-ART IFN study commenced after a median (IQR) of 175 days (112-217) of ART. Within 12 weeks of ART a transient increase in HCV RNA was seen in 18 of 19 patients, followed by a decrease such that the time-zero HCV RNA was lower in the post-ART study by a median (IQR) of 0.21 log10 IU/mL (0.06-0.56; p=0.002). Both pre- and post-ART, the HCV RNA nadir occurred 48 to 72 hours after IFN. The IFN response pre- and post-ART were closely correlated (at 72 hours: r=0.87; p<0.001) and both were associated with IL28B genotype (before p=0.02 and after p=0.005). The IFN response was not associated with baseline HIV RNA level, CD4+ T cell count, or T cell recovery. As hypothesized, the IFN response post-ART was greater than pre-ART; the difference was small and only significant at 72 hours (median (IQR) 0.11 log10 IU/mL; 0.00-0.40; p<0.05). Intrahepatic IFIT1 levels were inversely correlated with IFN response (r=-0.78; p<0.001) and its improvement post-ART (r=-0.56; p<0.05). While these data support the preference to begin ART before IFN based HCV treatment, the small improvement in early IFN response might also support withholding ART when interactions between HCV protease inhibitors and ART cannot safely be overcome.

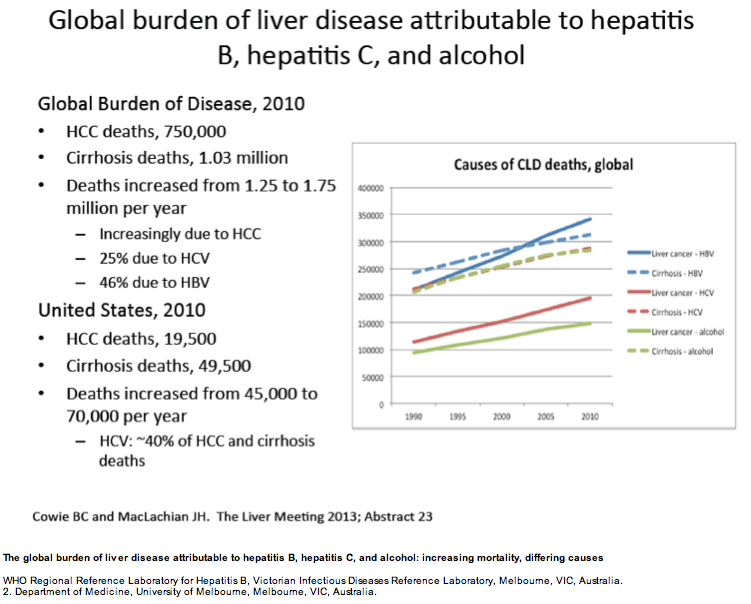
ABSTRACT BODY: Purpose: Establishing the relative contributions of the underlying causes of liver cancer and cirrhosis deaths is essential for appropriate targeting of public health and clinical responses at global, regional and national levels.
Methods: Using country-level and regional cause of death data released by the Global Burden of Disease Study 2010 (GBD 2010), we analysed the proportion of cirrhosis and liver cancer deaths attributable to HBV, HCV, alcohol, and other causes globally, but also in the USA, Western Europe, India, China and Australia.
Results: According to GBD 2010 data, there were an estimated 752 000 deaths from liver cancer and 1.03 million deaths from cirrhosis in 2010, making chronic liver disease a leading cause of human mortality. In the USA in 2010, approximately 70,000 people are estimated to have died from these causes. On a global basis, HBV is estimated to be responsible for 45% of liver cancer deaths and 30% of cirrhosis deaths, and HCV for 26% and 28%, respectively. Alcohol abuse is estimated to be responsible for approximately one quarter of liver cancer and cirrhosis deaths. With respect to regional estimates, HCV was the predominant cause of liver cancer/cirrhosis deaths in the USA (40/41%) and Western Europe (36/40%), with HBV predominating in China (54/46%) and India (48/35%). In Australia, leading causes of liver cancer and cirrhosis deaths were discordant; HBV lead as a cause of liver cancer deaths (41%) but alcohol was the predominant cause of cirrhosis deaths (33%).
Conclusions: These data from the GBD 2010 indicate that liver cancer and cirrhosis result in the deaths of 1.75 million humans each year, with chronic viral hepatitis causing approximately three quarters of these deaths. This analysis is drawn from a single study - the GBD 2010 - and must be considered together with other available data on causes of chronic liver disease worldwide. However the ability to systematically assess causes of disease and death using the same methodology across all regions and countries is an essential strength of GBD 2010. Greater priority to chronic viral hepatitis and other causes of liver disease is clearly needed to address this large burden of disease and death. The differing predominant causes of chronic liver disease identified across different regions requires prioritisation of prevention responses to address this emergent global health priority.
|
| |
|
|
|
|
|