| |
Targeting Innate HBV Immunity: A New Step in the Development of Combination Therapy for Chronic Hepatitis B; GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees
|
| |
| |
Download the PDF here
Download the PDF here
Gastroenterology June 2013
FABIEN ZOULIM
SOUPHALONE LUANGSAY
DAVID DURANTEL
INSERM U1052
Hospices Civils de Lyon and
Lyon University
Lyon, France
See "GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees," by Lanford RE, Guerra B, Chavez D, et al, on page 1508.
"In conclusion, the results presented by Lanford et al5 describing a novel approach to inhibit HBV replication and induce infected cell clearance provide refreshing perspectives on how to tackle chronic HBV infection (Figure 1). It represents a step toward the development of true combination therapy in chronic hepatitis B; with the aim of increasing the rate of serum HBsAg clearance."
Current treatments for chronic hepatitis B (HBV) based on nucleos(t)ide analogs (NUC) allow control of viral replication and liver disease in the majority of patients.1, 2 However, because NUCs are unable to clear viral covalently closed circular DNA (cccDNA), lifelong therapy is required to maintain the antiviral effect. To define new treatments with finite duration, it is therefore necessary to develop new molecules acting on novel targets to design true combination therapy complementing already existing NUC-based treatment.3 The persistence of HBV infection and the maintenance of the cccDNA mainly result from a weak HBV-specific immune response. In this respect, the stimulation of the immune response against HBV-infected cells might represent a relevant approach. An efficient control of viral infections requires a concerted action of both innate and adaptive immune responses4 (Figure 1). In the course of viral infections, the host surveillance system represents the first line of antiviral defense and rapidly recognizes viral nucleic acids and proteins. The activation of the innate immunity is essential for triggering an antiviral response of infected cells by producing type I and III interferons (IFNs), by inducing natural killer (NK) cell-mediated killing of viral infected cells, and by inducing the maturation and recruitment of adaptive immunity through production of pro-inflammatory cytokines and chemokines. In a second step, the adaptive immunity takes place after maturation and expansion of B- and T-cell clones that are able to specifically recognize the virus and/or infected cells. Altogether, the coordination of both branches of the immune response can lead to the control of viral infection.4 In this issue of Gastroenterology, Lanford et al5 have investigated the antiviral effect of the activation of the innate immunity via a Toll-Like receptor (TLR)-7 agonist, in HBV chronically infected chimpanzees.
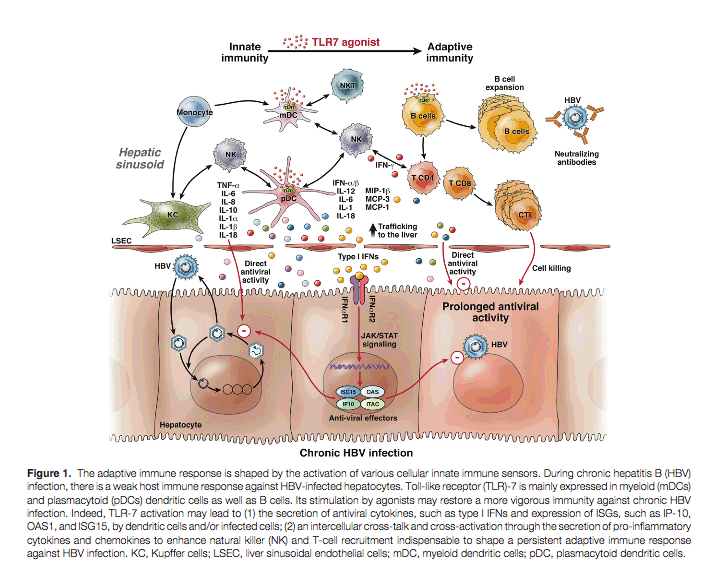
During viral replication, the production of many pathogen associated molecular patterns, such as intermediates of replication, that is, single- or double-strand RNA, viral DNA, core proteins or envelope glycoproteins, may be detected by innate immune sensors, namely, pathogen recognition receptors (PRRs), and trigger an antiviral response. In this respect, PRR activation serves as a link between innate and adaptive immunity. These receptors, mainly represented by the TLRs, the RIG-I like receptors, and the NOD-like receptors, may direct an effective antiviral response through the secretion of type I IFNs and pro-inflammatory cytokines.6 In the course of HBV infection, activation of innate responses may occur in the infected hepatocytes and in cells of the immune system, including circulating dendritic cells and NK cells, liver resident Kupffer cells, as well as other liver nonparenchymal cells.7, 8, 9 The current knowledge from both experimental and clinical studies suggest that HBV is a weak inducer of innate responses,10 but also exhibits a mechanism to suppress innate responses to establish a persistent infection of the liver.4 Indeed, it was shown that HBV may modulate innate responses by interfering either with TLR signaling pathways11 the type I IFN signaling pathway12, 13 or the cross-talk between plasmacytoid dendritic cells (pDC) and NK cells.14
TLR-7 is a PRR mainly expressed in endosomal compartments of pDCs and B lymphocytes that recognizes a pathogen-associated molecular pattern in viral single-stranded RNA. Upon stimulation of TLR-7, pDCs produce IFN-α and other cytokines/chemokines and induce the activation of NK cells and activation of cytotoxic lymphocytes, thereby orchestrating both innate and adaptive immune responses6 (Figure 1). The altered responsiveness of pDCs might contribute to the reduced innate immune responses during chronic viral infections.
Agonist-induced activation of TLR-7 might therefore represent a novel approach for the treatment of chronic viral infections.15
Lanford et al5 have investigated the effects of immune activation with GS-9620, an orally administered agonist of TLR-7, in chimpanzees chronically infected with HBV. GS-9620 was administered to chimpanzees every other day for 4 weeks at 1 mg/kg and, after a 1-week rest, for a second cycle of 4 weeks at 2 mg/kg. A detailed evaluation was performed, including pharmacokinetics of GS-9620, viral load, IFN-stimulated gene (ISG) expression, cytokine and chemokine levels, and lymphocyte and NK cell activation, as well as safety and tolerability parameters. Short-term administration of the TLR-7 agonist provided long-term suppression of serum and liver HBV DNA. The mean maximum reduction of viral DNA was 2.2 logs and occurred within 1 week of the end of therapy; reductions of >1 log persisted for several months. Serum levels of hepatitis B surface antigen and hepatitis B e antigen, and numbers of HBV antigen-positive hepatocytes, were reduced as hepatocyte apoptosis increased. In parallel, GS-9620 administration induced the production of IFN-α and other cytokines and chemokines, up-regulated ISGs expression, and activated NK cells and lymphocyte subsets, confirming the activation of TLR-7 signaling.
This is a timely study; new antiviral targets are required to improve the rate of hepatitis B surface antigen loss in patients with chronic hepatitis B. Theoretically, the TLR-7 agonist-induced antiviral activity may complement the inhibition of viral DNA synthesis induced by NUCs.3 From the results presented in this work, one might envision several mechanisms by which the TLR-7 agonist administration might have led to the observed antiviral effect5 (Figure 1). The decrease of both intrahepatic and serum viral load as well as the number of hepatitis B core antigen-positive hepatocytes was concomitant with an increase in liver enzymes and immune cell infiltration, suggesting HBV-infected cell killing by specific CD8+ T-cell responses and NK cell responses. This seems to be consistent with the role of infected cell killing and hepatocyte turnover in the subsequent clearance of infection.16 The determination of the respective role of hepatocyte apoptosis and/or lysis will also be important to fully understand the mechanism involved in the viral clearance process induced by this TLR-7 agonist. The induction of type-I IFN and ISGs expression may also have contributed to enhance the initial antiviral effect, similarly to the T helper 1 cytokine expression observed previously in experiments performed with HBV transgenic mice.17However, it is unlikely involved in the prolonged effect observed in these chimpanzees, because the induction of gene expression was only transient. It would also have been interesting to investigate the effect of this treatment regimen on the evolution of cccDNA amounts and its epigenetic control; it was previously shown in liver-humanized mice infected with HBV that IFN-α treatment induced the silencing of cccDNA by epigenetic reprogrammation18 and that immune-mediated killing of infected hepatocytes would lead to cccDNA decay.16 The determination of both integrated viral DNA and cccDNA would also have been extremely useful to ascribe the eventual cccDNA decay to the loss of infected cells or to infected cell curing.16 It will be also important to determine whether the positive effects observed in these infected chimpanzees receiving TLR-7 agonist monotherapy is confirmed, or even enhanced, in the context of combination therapy in chronically infected animals/patients who achieve viral DNA suppression in serum during NUC-based therapy.
One concern with TLR-7 agonist administration is the potential induction of adverse events, as previously observed with several other agonists evaluated in clinical trials for the treatment of chronic hepatitis C.19, 20 Flu-like syndromes and lymphopenia were observed in these clinical trials for hepatitis C.19, 20 The use of GS-9620 in HBV-infected chimpanzees was associated with lymphopenia as well as liver enzyme elevations; it is, however, interesting that there was no systemic production of IFN-α.5 Other potential adverse events resulting from an unregulated activation of TLR may include chronic inflammatory diseases and/or auto-immunity.6 Ongoing clinical trials will determine whether the administration of this specific TLR-7 agonist will provide the expected antiviral effect without the systemic effect associated with cytokine induction.
A better knowledge of the precise mechanism involved in HBV-induced repression of the infected host innate immunity might also help to design novel approaches to specifically trigger an antiviral innate response restricted to infected cells, with the caveat that the adaptive immune responses may not be involved in such a strategies.
In conclusion, the results presented by Lanford et al5 describing a novel approach to inhibit HBV replication and induce infected cell clearance provide refreshing perspectives on how to tackle chronic HBV infection (Figure 1). It represents a step toward the development of true combination therapy in chronic hepatitis B; with the aim of increasing the rate of serum HBsAg clearance.
|
|
| |
| |
|
|
|