| |
A phase 2B study of MK-7009 (vaniprevir) in patients with genotype 1 HCV
infection who have failed previous pegylated interferon and ribavirin treatment
|
| |
| |
Download the PDF
Journal of Hepatology July 2013
Eric Lawitz1, Maribel Rodriguez-Torres2, Albrecht Stoehr3, Edward J. Gane4, Lawrence Serfaty5, Sanhita Bhanja6, Richard J. Barnard6, Di An6, Jacqueline Gress6, Peggy Hwang6, Niloufar Mobashery6,
1Alamo Medical Research, San Antonio, TX, United States; 2Fundacion de Investigacion, San Juan, Puerto Rico; 3IFI - Institut fur Interdisziplinare
Medizin, Hamburg, Germany; 4Auckland Clinical Studies Ltd, Auckland, New Zealand; 5Service d'Hepatologie, Hopital Saint-Antoine, Universite
Pierre et Marie Curie, Paris, France; 6Merck Sharp & Dohme, Whitehouse Station, NJ, United States
MK-7009 therapy at 300 mg and 600 mg b.i.d. was generally well tolerated for up to 48 weeks of therapy. Patients receiving MK-7009 regimens had higher rates of gastrointestinal (GI) adverse events (AEs) when compared with control. Most GI AEs were mild to moderate in severity. There were no significant differences in rates of anemia and rash between the patients receiving MK-7009 and control.
In conclusion, patients treated with MK-7009 plus P/R experienced significant improvement in SVR compared to P/R control in a population of GT 1 experienced patients. Results show that MK-7009 compares favorably with other first generation protease inhibitors. MK-7009 was generally well tolerated and most adverse experiences were mild to moderate in severity. Based on these findings, the development of MK-7009 has advanced to phase 3 in Japan.
Background & Aims
MK-7009 (vaniprevir) is a non-covalent competitive inhibitor of the hepatitis C virus (HCV) NS3/4A protease. This report presents the primary analysis results (safety and sustained viral response) of a phase 2b study of MK-7009 given in combination with peginterferon (PegIFN) alfa2a 180 μg weekly and ribavirin (RBV) 1000-1200 mg/day, for 24-48 weeks to non-cirrhotic patients who have failed previous PegIFN and RBV treatment.
Methods
We present results of a randomized, placebo-controlled, double-blind study of MK-7009 administered for 24-48 weeks in combination with PegIFN and RBV in 4 regimens to at least 40 patients per arm. Stratification by prior response to PegIFN and RBV was as follows: null response, partial response, breakthrough and relapse. HCV RNA was determined by Roche Cobas Taqman with a lower limit of detection (LLoD) of 10 IU/ml and a lower limit of quantification (LLoQ) of 25 IU/ml.
Results
SVR24 in patients on MK-7009 + PegIFN and ribavirin (P/R) was statistically superior to placebo + P/R in all treatment groups (p <0.001). MK-7009 at 300 mg b.i.d. and 600 mg b.i.d. is generally well tolerated for use for up to 48 weeks of therapy. Patients in MK-7009 regimens had higher rates of gastrointestinal adverse events as compared to control (mostly mild to moderate). There were no significant differences in rates of anemia and rash between the MK-7009 regimens and control.
Conclusions
In conclusion, patients treated with MK-7009 plus P/R experienced significant improvement in SVR compared to P/R control in a population of GT 1 experienced patients.
Introduction
Hepatitis C virus (HCV) is a positive-strand RNA virus of the Flaviviridae family and replicates primarily in the liver. While disease progression is typically a slow process that occurs over many years, a significant fraction of patients ultimately develop serious liver disease, including cirrhosis and hepatocellular carcinoma [1].
Multiple viral proteins essential for HCV replication have been characterized [1], [2] and clinical proof of concept has been demonstrated for small-molecule inhibitors that act against several of these, including NS3/4A protease [3], [4], NS5B polymerase (both active site and allosteric inhibitors) [5], [6], [7], [8], NS4A [9], and most recently, NS5A [10]. Of these, NS3/4A protease inhibitors have progressed the furthest in terms of clinical evaluation and have been demonstrated to achieve highly significant reductions in HCV viral loads in patients [11].
Until recently, standard treatment for HCV infection was combination therapy with pegylated interferon and ribavirin (P/R) [12], [13], [14]. However, recent FDA approval of the directly acting antiviral agents boceprevir [15], [16] and telaprevir [17], [18] has added to the HCV treatment armamentarium. These compounds have been added to the previous P/R backbone in genotype 1 infected patients to create a 3-drug treatment regimen. Both boceprevir and telaprevir are orally bioavailable α ketoamide NS3/4A protease inhibitors that reversibly and covalently bind to the HCV protease. A sustained virological response (SVR) of up to 70% is observed when either of these protease inhibitors is utilized with P/R and ribavirin in treatment naive patients [19]. Both agents are also effective in patients who failed to achieve SVR during previous treatment with P/R, though overall SVR rates are generally lower.
In addition to the already approved HCV protease inhibitors, there are several other investigational second-wave NS3/4A protease inhibitors. Vaniprevir (MK-7009) is one such compound and is a macrocyclic HCV NS3/4A protease inhibitor which has demonstrated potent antiviral efficacy and good tolerability in a 14-day phase I monotherapy trial as well as a phase II dose-ranging study [20].
This manuscript presents the primary analysis results (safety and sustained viral response) of a phase 2b study of MK-7009 given in combination with PegIFN 180 μg weekly and RBV 1000-1200 mg/day for 24-48 weeks, to non-cirrhotic patients who have failed previous P/R treatment.
Materials and methods
Study design
MK-7009 safety and efficacy had been established in a phase 2A study of treatment na�ve patients (MK-7009 study 007) [20]. The data presented in this report are from a randomized, placebo-controlled, double-blind study of MK-7009 administered for 24 or 48 weeks to non-cirrhotic patients, who failed previous P/R treatment, enrolled from Australia, Austria, Belgium, Canada, Chile, Czech Republic, France, Germany, Israel, Korea, Lithuania, New Zealand, Poland, South Korea, Sweden, Taiwan, Thailand, UK, and the USA. Patients were treated with MK-7009 for longer durations in the current study when compared to MK-7009 study 007 with the assumption that the demonstration of safety and efficacy in a treatment experienced population would imply safety and efficacy in a treatment na�ve population. The study was conducted in accordance with principles of Good Clinical Practice and the Declaration of Helsinki and was approved by the appropriate institutional review boards and regulatory agencies. Informed consent was documented for each patient prior to study enrollment.
Patient safety was overseen by an external Data Monitoring Committee made up of 4 internationally recognized hepatologists and/or infectious disease specialists and one statistician. The committee met quarterly at a minimum to discuss safety and efficacy. Additionally, direct patient monitoring for safety by principal investigators included patient history, physical exams and pre-specified laboratory safety evaluations at study visits on days 1, 3 and 7 for the first week, weekly from weeks 2 through 4, every other week from weeks 5 through 12 and then monthly through end of study treatment.
MK-7009 was administered in combination with P/R in 4 regimens (one 300 mg b.i.d. regimen and three 600 mg b.i.d. regimens) to at least 40 patients per arm of the study (Fig. 1A). There was also a control arm that received placebo + P/R. Patients were stratified based on prior response to P/R (null response, partial response, breakthrough and relapse). Key inclusion criteria for entry into the study included chronic genotype (GT) 1 HCV-infected patients who have failed prior treatment(s) with P/R, a minimum of 25% of patients prior null responders, men and women 18-65 years of age, and baseline HCV RNA 4 x 105 IU/ml. Key exclusion criteria included non-HCV-related chronic hepatitis, HIV co-infection, evidence of cirrhosis on liver biopsy or approved non-invasive imaging, or any other condition contraindicated for treatment with P/R.
HCV RNA was measured at every scheduled study visit and detected by Roche Cobas Taqman. This assay has a lower limit of detection (LLoD) of 10 IU/ml and a lower limit of quantification (LLoQ) of 25 IU/ml.
Objectives
This study's primary safety objective was to evaluate the safety and tolerability of the MK-7009 300 mg b.i.d. treatment regimen and the three MK-7009 600 mg b.i.d. treatment regimens as compared with placebo in combination with 48 weeks of P/R (hereafter referred to as the control regimen), as assessed by review of the accumulated safety data.
The primary efficacy objective was to evaluate the antiviral activity of the three MK-7009 600 mg b.i.d. treatment regimens as compared with the control regimen, as assessed by the proportion of patients achieving undetectable HCV RNA 24 weeks after the end of all study therapy (sustained viral response 24 [SVR24]).
Secondary objectives included: (1) to evaluate the antiviral activity of the MK-7009 300 mg b.i.d. treatment regimen as compared with the control regimen, as assessed by the proportion of patients achieving SVR24; (2) to evaluate the antiviral activity of treatment regimen 1 (24 weeks of MK-7009 600 mg b.i.d. in combination with P/R) as assessed by the proportion of patients achieving SVR24, as compared with response at treatment week 48 in the control regimen; (3) to evaluate the antiviral activity of MK-7009 300 mg b.i.d. treatment regimen and two MK-7009 600 mg b.i.d. treatment regimens (treatment regimens 2, 3, and 4) as compared with the control regimen, as assessed by the proportion of patients achieving undetectable viral RNA at treatment week 48.
Data analysis
Safety analyses are based on 'all patients as treated' (APaT) population. The full analysis set (FAS) population served as the primary population for the efficacy analysis. This population included all randomized patients who received at least one dose of study medication and had post-dose data. For this study, there is no difference in the results whether we use the FAS population or the Intent-to-Treat (ITT) population. Although we required the patients to have at least one post-randomization end point subsequent to at least one dose of study treatment to be included in the FAS population, every patient who was randomized and received at least one dose of study medication had at least one post-randomization HCV RNA measurement. Therefore, no patient was excluded based on the requirement of a post-randomization end point and the FAS population was identical to the ITT population.
With a sample size of ~40 per treatment arm, there was 82% power to demonstrate that at least one of the MK-7009 treatment regimens was superior to the control regimen as assessed by SVR24. This assumes a true difference in SVR24 of 32% at the point of maximum variability for a binary end point (for instance, SVR24 of 66% for the MK-7009 treatment regimens and an SVR24 of 34% for the control regimen; the maximum variability for a binary end point is 50%).
Criteria for virologic failure
HCV RNA data was provided to the designated unblinded Clinical Monitor and patients were discontinued from all study therapy if they met any of the following criteria for virologic failure: if the patient did not achieve at least a 2-log10 IU/ml decline in HCV RNA from baseline by treatment week 12; if the patient had quantifiable HCV RNA at treatment week 24 (if the patient had positive but non-quantifiable HCV RNA at treatment week 24, a second measurement was taken at treatment week 28, if applicable; if the week 28 result was also positive, the patient was discontinued from the study); if the patient had evidence of breakthrough viremia, as defined by: (1) a >1-log10 increase from nadir viral RNA (nadir calculated from up to two consecutive HCV RNA measurements) in two consecutive visits by week 12, or (2) a plasma viral RNA of >100 IU/ml, in two consecutive visits after becoming undetectable; if the patient had evidence of relapse, as defined by two consecutive visits with detectable HCV RNA following the end of all study treatment after becoming undetectable on treatment.
Results
Overall, 211 patients were enrolled into the five treatment regimens (Fig. 1B). Of those, 62.1% were male (n = 131) and 37.9% were female (n = 80) (Table 1). Overall, 190 patients (90.0%) completed the study, and 20 (9.5%) discontinued (Supplementary Table 1). The most common reasons for study discontinuation were 'withdrawal by subject', and 'lost to follow-up'. The mean age of enrolled patients was 49.6 years, and age ranged from 23 to 65 years. Caucasian patients made up the majority of the population (77.7%), followed by Asian (13.7%) and Black/African American (7.1%) patients. The population was skewed slightly towards HCV 1b infection (57.3%); 41.7% of patients were infected with HCV 1a. Relapse made up the largest portion of prior failures (39.3%), followed by null response (25.6%), partial response (19.9%) and breakthrough (15.2%). Of the patients with IL28B SNP data available, 26 (19.8%) and 105 (80.2%) were CC and CT/TT, respectively.
SVR24 rates in the ITT population can be seen in Table 2. Patients given 24 or 48 week courses of MK-7009 600 mg b.i.d. + P/R had SVR24 rates of 71.1% and 78.0%, respectively. While there may be no obvious advantage to a longer treatment regimen, the study was not powered to discern differences between 24- and 48-week treatments. The SVR24 rate in patients randomized to the 300 mg b.i.d. + P/R treatment regimen was 66.7%. The largest percentage of patients achieving SVR24 (84.2%) was seen in the 24-wk 600 mg bid + P/R/24-wk PBO + P/R treatment group. SVR24 in patients on MK-7009 + P/R was statistically superior to placebo + P/R in all treatment groups (p <0.001). Thirteen patients in the vaniprevir treatment groups were excluded from the primary analysis based on the pre-specified missing data approach. Patients who discontinued for administrative reasons, whose last HCV RNA measurement was undetectable prior to discontinuation, were not included. Patients who discontinued for treatment-related reasons, regardless of HCV RNA measurements, were considered failures. Reasons for virologic failure are listed in Supplementary Table 2.
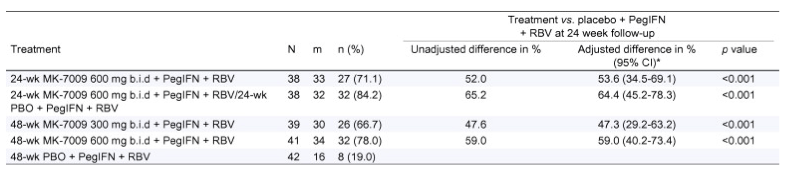
*The adjusted difference is the difference of the two proportions (MK-7009 treatment regimen minus control regimen) adjusting for the stratification variable with Cochran Mantel-Haenszel (CMH) weights. The Miettinen and Nurminen method was used for this analysis.
N, number of patients included in the analysis; m, number of patients with an HCV RNA result at the 24 week follow-up visit; n (%) = number of patients with undetectable HCV RNA at the 24 week follow-up visit and the percentage calculated as (n/N) * 100; CI, confidence interval; ITT, intention to treat.
SVR24 rates by prior response to therapy reveal that the highest response rates were seen in patients who previously either partially responded or relapsed (Table 3A). Importantly, the results of this analysis are based on small numbers due to the stratification into several patient groups. When SVR24 rates were examined by HCV genotype, there were no obvious differences in response to two of the three MK-7009 treatment regimens between genotype 1a and 1b (Table 3B). Though the numbers are small, a difference that is evident between genotype 1a and genotype 1b is the response in patients given 24 weeks of treatment with MK-7009 600 mg b.i.d. SVR24 in this population was 56.3% in patients with type 1a and 81.8% in patients with type 1b. However, no conclusive statements can be made as the study was not powered to study genotype subtype differences. Similarly, the samples sizes become small as the SVR rates are examined by baseline HCV RNA levels and fibrosis staging; thus the data were not instructive (data not shown).
Table 3. SVR24 rates by (A) prior response to therapy and (B) genotype in the ITT population
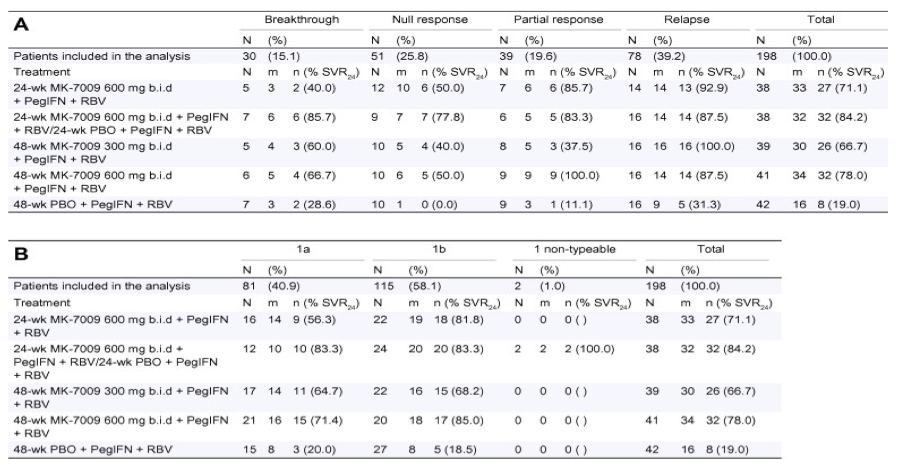
(A) N, number of patients included in the analysis by prior treatment response; (%), percentage of patients included in the analysis calculated as (prior treatment response N/total N) * 100; m, number of patients with an HCV RNA result at the 24 week follow-up visit; n (% SVR24), number of patients with undetectable HCV RNA at the 24 week follow-up visit and the percentage calculated as (n/N) * 100.
(B) N, number of patients included in the analysis by HCV genotype; (%), percentage of patients included in the analysis calculated as (HCV genotype N/total N) * 100; m, number of patients with an HCV RNA result at the 24 week follow-up visit; n (% SVR24), number of patients with undetectable HCV RNA at the 24 week follow-up visit and the percentage calculated as (n/N) * 100.
IL28B status was collected from 131 patients who provided genetic consent and were analyzed in the per-protocol analyses. Of these, 19.8% (26/131) were CC, 59.5% were CT (78/131) and 20.6% were TT (27/131) for the SNP rs12979860. While IL28 genotype interactions were seen with SVR24 when considering all treatment groups, the effect of IL28 genotype became non-significant when considering only the MK-7009 groups (data not shown). Therefore it appears that the interaction was mainly driven by the difference in actual SVR24 rates between PBO and MK-7009 treatment arms. Moreover, it was found that for each MK-7009 treatment arm, there was no significant evidence that CC genotype patients achieved SVR24 at a higher rate (Fig. 2).
In total, 83.3% of patients in the PBO arm and between 90.0% and 97.8% of patients in the MK-7009 treatment arms reported one or more drug-related adverse events during the treatment period (Table 4). Overall, between 5 and 8 patients in each of the MK-7009 treatment arms and 7 patients in the PBO arm had reported anemia. To address the anemia, 11 patients received epoetin beta during the trial. There were no significant differences in rates of anemia and rash between the MK-7009 regimens and the control group. No serious rashes were reported. Patients in MK-7009 regimens had higher rates of gastrointestinal adverse events as compared to control (Table 4). A complete adverse event summary is reported in Supplementary Table 3. The most common gastrointestinal adverse events reported were diarrhea, nausea, and vomiting. Most gastrointestinal adverse experiences were mild to moderate in intensity; few were severe (Supplementary Table 4). Other adverse events with incidences 25% are shown in Supplementary Table 5.
Table 4. Adverse events of clinical interest during the treatment phase.
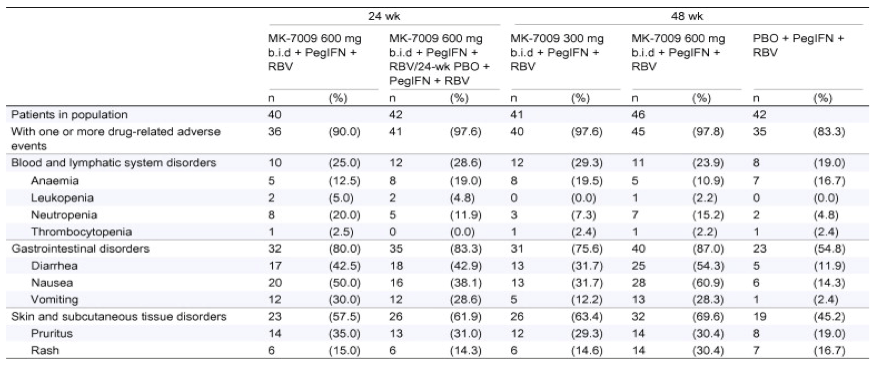
Every patient is counted a single time for each applicable row and column.
A system organ class or specific adverse event appears on this report only if its incidence in one or more of the columns meets the incidence criterion in the report title, after rounding.
Discussion
First generation HCV protease inhibitors such as boceprevir and telaprevir have strong antiviral efficacy, but need to be dosed frequently (8 or 12 h) and can be associated with anemia, dysgeusia, and skin rashes [21], [22]. MK-7009 is a macrocyclic HCV NS3/4A protease inhibitor (administered QD or BID) that has demonstrated strong antiviral potency and a good safety profile in phase I studies [23].
In this report, MK-7009 (when combined with pegylated interferon alfa 2a and ribavirin) produced a significant improvement in the rate of SVR compared to retreatment with P/R in patients who had failed previous therapy for chronic hepatitis C. The highest rates of SVR24 were seen in patients who relapsed after initial P/R treatment; where cumulatively MK-7009 treated patients had an SVR24 of 94%. This result may be in part the result of the higher number of patients with genotype 1b, although the overall results appear to demonstrate no significant difference. The outcomes of this trial compare favorably to the trials of telaprevir and boceprevir [24], [25]. In this trial, prior null responder patients had numerically higher SVR point estimates compared to the data from telaprevir- and boceprevir-based therapies, although direct comparisons of these data to previous data for boceprevir and telaprevir are precluded due to lack of head-to-head studies. Observed MK-7009 SVR24 rates are similar to another 'first generation second wave' HCV protease inhibitor, TMC-435 [26]. Cirrhotic patients were randomized in this study in a separate cohort in a sequential fashion; the results will be analyzed once all cirrhotic patients have completed the follow-up visits.
Resistance is an important consideration in the use of HCV protease inhibitors [27]. Resistance-associated amino acid variants (RAVs) in response to MK-7009 therapy were predominantly observed at positions R155, A156 and/or D168 in non-SVR patients. This resistance profile is similar to other first generation NS3/4 inhibitors [28], [29], [30], [31].
MK-7009 therapy at 300 mg and 600 mg b.i.d. was generally well tolerated for up to 48 weeks of therapy. Patients receiving MK-7009 regimens had higher rates of gastrointestinal (GI) adverse events (AEs) when compared with control. Most GI AEs were mild to moderate in severity. There were no significant differences in rates of anemia and rash between the patients receiving MK-7009 and control.
In conclusion, patients treated with MK-7009 plus P/R experienced significant improvement in SVR compared to P/R control in a population of GT 1 experienced patients. Results show that MK-7009 compares favorably with other first generation protease inhibitors. MK-7009 was generally well tolerated and most adverse experiences were mild to moderate in severity. Based on these findings, the development of MK-7009 has advanced to phase 3 in Japan.
|
|
| |
| |
|
|
|