| |
the FDA & the Janssen Briefing Documents Oct 24 Hearing
|
| |
| |
Download the PDF here
Download the PDF here
Download the PDF here
3 pdfs attached
FDA ANTIVIRAL DRUGS ADVISORY COMMITTEE MEETING
OCTOBER 24, 2013
BACKGROUND PACKAGE
FOR
NDA 205123
SIMEPREVIR (TMC435)
FOOD AND DRUG ADMINISTRATION
CENTER FOR DRUG EVALUATION RESEARCH
OFFICE OF ANTIMICROBIAL PRODUCTS
DIVISION OF ANTIVIRAL PRODUCTS
Background Package for NDA 205123
"Based on the above analyses, as well as our experience with the approved NS3/4A protease inhibitors, DAVP believes that it is reasonable to extend the indication of simeprevir for the treatment of partial and null responders. Patients in these populations would receive 12 weeks of simeprevir in combination with PR, followed by an additional 36 weeks of PR. DAVP believes screening for the presence of the Q80K polymorphism (as discussed in detail above) will be especially important in these populations in which SVR is already anticipated to be more difficult to achieve. DAVP intends to designate the submission of the final data from the ongoing Phase 3 trial of simeprevir in combination with PR in partial and null responders (HPC3001) a post-marketing commitment."
2.7 General Summary:
In the pivotal Phase 3 trials, C208, C216, and HPC3007, simeprevir in combination with PR was demonstrated to be superior to placebo (in combination with PR) in achieving a sustained virologic response in both HCV treatment-naïve subjects and subjects who relapsed after prior pegylated interferon-ribavirin therapy. In the subgroup of subjects with the Q80K baseline polymorphism, a substantial impact on the efficacy of simeprevir was observed. Given the high frequency of the Q80K polymorphism in the U.S. population and its significant impact on rates of SVR12, DAVP is recommending that all GT1a patients undergo screening for this baseline polymorphism prior to treatment with simeprevir and that alternative treatment options be considered for patients found to be infected with this polymorphic variant. DAVP believes that it is reasonable (in the setting of a Q80K screening recommendation) to extend the indication of simeprevir for the treatment of partial and null responders based on the available data, including the results of trial C206, a Phase 2b trial which included these patient populations.
Subjects with moderate or severe hepatic impairment and subjects of East Asian ancestry had substantial increases in mean simeprevir exposures compared to healthy subjects and compared to the pooled Phase 3 population, respectively. Based on 1) the paucity of safety data in a subjects having mean simeprevir exposures 2- to 5-fold higher than the mean observed in Phase 3 trials; and 2) the positive relationship between simeprevir exposures and the incidence of adverse events including rash and photosensitivity, DAVP recommends a reduced simeprevir dose for patients with moderate or severe hepatic impairment or patients of East Asian ancestry. However, as no reduced dose strengths are currently available, definitive dose recommendations and labeling for these populations will likely need to be accomplished as postmarketing requirements or commitments.
The nonclinical safety package for simeprevir was deemed adequate with the liver and GI system identified as the major target organs across animal species. Simeprevir will be categorized as a Pregnancy Category C in labeling based on reproductive toxicities in the rat and mouse that indicate potential adverse effects in the pregnant animal, the fetus, and the developing offspring.
The safety profile of simeprevir was generally acceptable. An increased frequency and severity of hyperbilirubinemia was associated with simeprevir use, however no association between the bilirubin elevation and clinically relevant hepatototoxicty was appreciated. The major safety signal identified in the review involved rash and/or photosensitivity events. This included an increased frequency and severity of rash/photosensitivity adverse events and serious adverse events, as well as an increase in rates of discontinuation of simeprevir due to rash/photosensitivity related adverse events. DAVP intends to include a warning related to photosensitivity in the prescribing information including a recommendation for sun protection measures for all patients receiving simeprevir.
3.0 PRELIMINARY TOPICS FOR THE ADVISORY COMMITTEE
The Division is convening this meeting to solicit the committee's comments on the following topics. Please note, however, that these are preliminary topics and are still subject to change.
1. Please comment on the safety profile of simeprevir focusing on rash and photosensitivity events reported during the clinical trials.
a. Does the committee agree that a discussion of the photosensitivity events should be included in the Warnings and Precautions section of the simeprevir prescribing information?
b. Based on the available data, does the committee agree that sun-protection measures should be recommended for all patients receiving simeprevir?
c. Does the committee believe it appropriate and/or necessary to include a discussion of rash events (separate from that for photosensitivity) in the Warnings and Precautions section of the prescribing information?
2. Considering the overall risks and benefits, do the available data support approval of simeprevir in combination with pegylated interferon and ribavirin for treatment of HCV infection?
a. DAVP intends to recommend screening all subjects with GT1a infection for the Q80K polymorphism prior to initiation of simeprevir (in combination with pegylated interferon and ribavirin) and that alternative treatment options be considered for patients with this baseline polymorphism. Does the committee agree with DAVP's proposed approach to managing the reduction in efficacy apparent in the setting of the Q80K polymorphism?
3. At the proposed dose of simeprevir 150 mg once daily, mean exposures were approximately 3.4-fold higher in individuals of East Asian ancestry compared to the pooled Phase 3 population. Similarly, simeprevir 150 mg once daily provided 2.4- and 5.2-fold higher exposures in subjects with moderate or severe hepatic impairment, respectively, compared to healthy controls. Considering the lack of safety data in patients with mean exposures that are 2- to 5-fold higher compared to those observed in the Phase 3 population, as well as the positive relationship between simeprevir exposures and the incidence of adverse events (including rash, photosensitivity, pruritus, dyspnea, and increased bilirubin), should the dose strength of simeprevir be reduced in the following patient subgroups:
a. Patients of East Asian ancestry
b. Patients with moderate or severe hepatic insufficiency
4. Are there post marketing studies that should be conducted to further define risks or to optimize use of simeprevir?
DISCLAIMER STATEMENT
The attached package contains background information prepared by the Food and Drug Administration (FDA) for the panel members of the advisory committee. The FDA background package often contains assessments and/or conclusions and recommendations written by individual FDA reviewers. Such conclusions and recommendations do not necessarily represent the final position of the individual reviewers, nor do they necessarily represent the final position of the Review Division or Office. We have brought simeprevir to this Advisory Committee in order to gain the Committee's insights and opinions, and the background package may not include all issues relevant to the final regulatory recommendation and instead is intended to focus on issues identified by the Agency for discussion by the advisory committee. The FDA will not issue a final determination on the issues at hand until input from the advisory committee process has been considered and all reviews have been finalized. The final determination may be affected by issues not discussed at the advisory committee meeting.
1.0 REGULATORY BACKGROUND AND INTRODUCTION
The purpose of this document is to provide the Antiviral Products Advisory Committee (AVAC) with a summary of FDA analyses of data submitted by Janssen Research and Development, LLC in support of simeprevir (TMC435) for the treatment of chronic hepatitis C genotype-1 (GT1) infection, in combination with peginterferon-alpha and ribavirin (PR), in adult patients with compensated liver disease, including cirrhosis, who are treatment-naïve or who have failed previous interferon-based therapy. During the Advisory Committee meeting to be held on October 24, 2013, the Committee will be asked to consider the safety and efficacy data submitted to support the approval of simeprevir for this indication. The background materials provided represent the findings and opinions of the primary reviewers from different disciplines, based on their reviews of the Applicant's submissions. It must be emphasized that this document represents the review team's preliminary findings, and that no regulatory decision has been made on the status of the application. Indeed, the advice the AVAC provides will be critical in our regulatory decision making.
Simeprevir is a specific inhibitor of the hepatitis C virus (HCV) NS3/4A serine protease. Hepatitis C virus (HCV) protease inhibitors block the NS3/4A protease-dependent cleavage of the HCV polyprotein, thereby inhibiting viral replication in infected host cells. If approved, simeprevir would represent the third HCV protease inhibitor approved in the US. The HCV protease inhibitors, boceprevir (Victrelis®) and telaprevir (IncivekTM) were initially approved in 2011.
The current application requests approval for simeprevir in combination with PR for the treatment of chronic HCV in adult patients who are treatment-naïve or who have failed previous interferon-based therapy. The proposed indication is supported by the 60-week safety and efficacy data from the phase 3 trials TMC435-TiDP16-C208, TMC435-TiDP16-C216, and TMC435HPC3007 (hereafter referred to as C208, C216, and HPC3007, respectively in our document) and the completed phase 2b trials TMC435-TiDP16-C205 and TMC435-TiDP16-C206 (hereafter referred to as C205 and C206 in our document). A safety update report was also provided in support of the current application.
2.0 CLINICAL AND NON-CLINICAL DATA
2.1 Study Designs:
The Phase 3 trials C208, C216, and HPC3007 were randomized, double-blind, placebo controlled trials in subjects chronically infected with HCV GT1. These trials assessed the combination of simeprevir (dosed at 150 mg daily for 12 weeks) plus PR for 12 weeks followed by PR alone for either 12 or 36 weeks based on an individual subject's virologic response to therapy (hereafter referred to as response guided therapy or RGT). The Control arm in each of these trials was placebo (for 12 weeks) in combination with PR for a fixed 48 week duration. The primary endpoint for these trials was sustained virologic response 12 weeks after the planned end of treatment (SVR12). SVR12 was defined as an undetectable HCV RNA at the end of treatment and HCV RNA < 25 IU/mL 12 weeks after the planned end of treatment. Stratification factors included HCV genotype/subtype and IL28B status. Trials C208 and C216 were identical in design and enrolled only treatment-naïve subjects. Trial HPC3007 enrolled subjects who had received at least 24 weeks of a pegylated interferon-based therapy and had relapsed within 1 year after the last medication intake (hereafter referred to as "relapsers" in our document).
Data from two Phase 2b trials, C205 and C206, were also provided in support of the proposed indication. Study C205 was a randomized, double-blind, 5-arm, placebo-controlled trial to investigate the safety and efficacy of simeprevir (dosed at 75 and 150 mg daily and given for either 12 or 24 weeks) in combination with PR (for 24 or 48 weeks based on RGT) in treatment-naïve CHC genotype-1 infected subjects. The Control arm was placebo (for 24 weeks) in combination with PR (for 48 weeks). The primary endpoint for this trial was SVR at week 72. This study served as an ancillary source of safety data for our review.
The C206 trial was a randomized, double-blind, 7-arm, placebo-controlled trial to compare the safety and efficacy of different regimens of simeprevir (100 or 150 mg daily) plus PR versus PR alone in CHC GT1- infected subjects who had failed to respond during or had relapsed following at least 1 course of PR therapy. Subjects in study C206 were randomized in a 1:1:1:1:1:1:1 fashion over 6 simeprevir dose groups and 1 placebo group (see Figure 1 for details). In treatment groups 1 and 4, subjects received 12 weeks of triple therapy with 100 or 150 mg simeprevir q.d. plus PR, followed by 36 weeks of treatment with PR and simeprevir-matched placebo (identified as the TMC12PR48 100 mg and TMC12PR48 150 mg groups in figure 1 respectively). In treatment groups 2 and 5, subjects received 24 weeks of triple therapy with 100 or 150 mg simeprevir q.d. plus PR (identified as TMC24PR48 100 mg and TMC24PR48 150 mg groups in figure 1 respectively). In treatment groups 3 and 6, subjects received 48 weeks of triple therapy with 100 or 150 mg simeprevir q.d. plus PR (identified as TMC48PR48 100 mg and TMC48PR48 150 mg groups in figure 1 respectively). In treatment group 7, subjects received PR and simeprevir-matched placebo for 48 weeks (Control group).
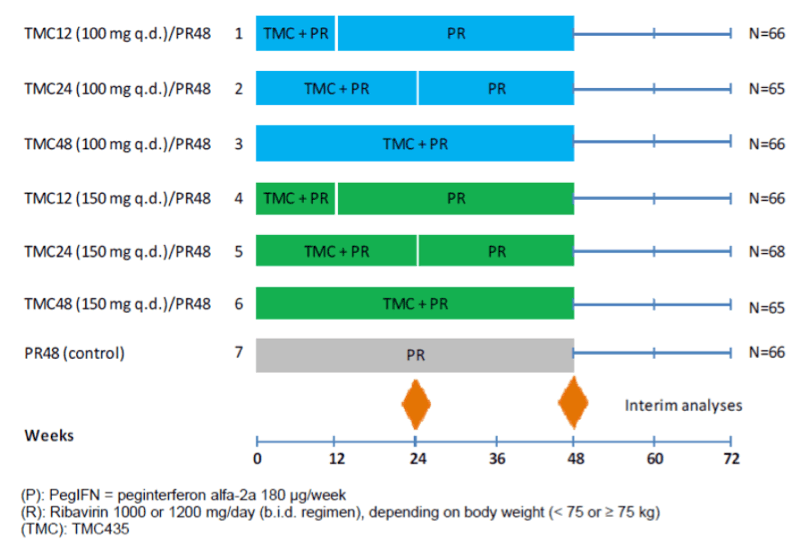
Data from the C206 trial was provided to support an indication in patients characterized as partial or null responders to previous pegylated interferon therapy. This study served as an ancillary source of safety and efficacy data for our review.
2.2 Demographics:
Phase 2b Trials
In the C205 trial, a total of 386 subjects were enrolled. Eleven percent of subjects were drawn from the Asia-pacific region, 68% from Europe and 21% from North America. In Study C206, a total of 462 subjects were enrolled. Sixty-eight percent of subjects were drawn from Europe, 6% from Australia/New Zealand, and 26% from North America.
Phase 3 Trials
In the C208 trial, a total of 394 patients were randomized from 13 countries. Fourteen percent of subjects were drawn from the Asia-pacific region, 42% from Europe and 44% from North America (with 30% from the United States). In C216, a total of 391 patients were randomized from 14 countries. Sixty-five percent of subjects were drawn from Europe, 15% from South America and 20% from North America (all from the United States). In HPC3007, a total of 393 patients were randomized from 14 countries. Eight percent of subjects were drawn from the Asia-pacific region, 70% from Europe and 22% from North America (with 18% from the United States). Demographic characteristics were generally well balanced between the simeprevir arms and Control arms for each of the Phase 3 trials (C208, C216, and HPC3007). The majority of subjects in all arms of the phase 3 trials were Caucasian (range 86-96%) and non-Hispanic/Latino (range 77-95%). Trial C208 had the highest representation of North American subjects at 44%, while rates in C216 (20%) and HPC3007 (22%) were substantially lower. Cirrhotic subjects (Metavir Fibrosis score of F4) comprised from 7 to 15% of subjects across study arms. IL28b CC status ranged from 24-31% across study arms. Both HCV genotype/subtype 1a and 1b subjects were well represented in the Phase 3 trials (range for 1a across arms: 40-57%; range for 1b across arms: 43-59%).
2.3 Clinical Efficacy Results:
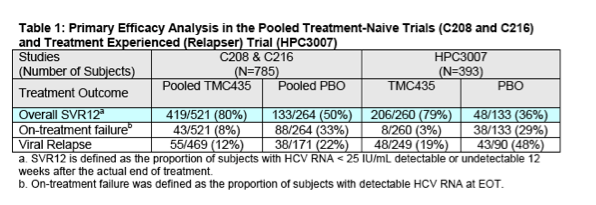
Primary Efficacy Endpoint: As discussed above, the Applicant's proposed indication for the treatment of chronic HCV infection is based primarily on the SVR12 results from the Phase 3 pivotal trials (C208, C216, and HPC3007). As trials C208 and C216 were both performed in a HCV treatment-naïve population and employed a nearly identical study design, efficacy results were pooled for analysis. The pooled SVR12 results from the treatment-naïve studies demonstrated an SVR12 rate of 80% in the simeprevir group and 50% in the Control group. Trial HPC3007 was performed in patients who had relapsed after previous pegylated interferon/ribavirin treatment for chronic hepatitis C infection. In HPC3007, the SVR12 rate in the simeprevir arm was 79% compared to 36% in the Control arm. Please refer to the Table 1 below for details.
Secondary Efficacy Endpoints: SVR24 and SVR72 were included as secondary endpoints in each of the phase 3 pivotal trials. Both SVR24 and SVR72 results correlated well with the primary SVR12 endpoint. However, SVR24 and SVR72 data were incomplete at the time of the Week 60 data cutoff.
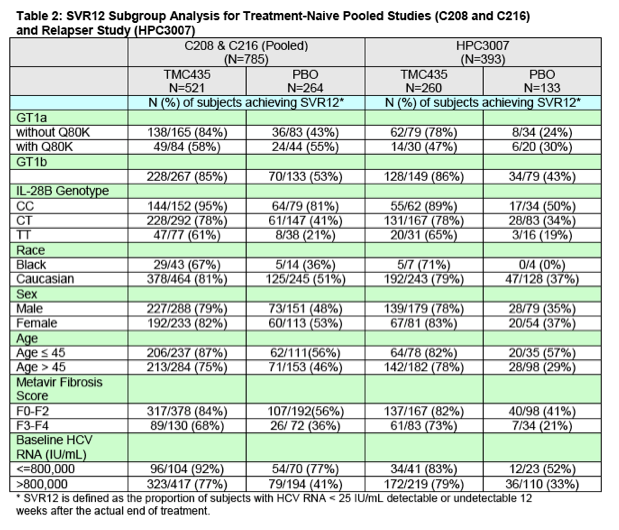
Subgroup Analyses: Table 2 presents SVR12 data by subgroups from the pooled studies in treatment-naïve subjects (C208 and C216) as well as from the trial in subjects who relapsed after prior interferon-based therapy (HPC3007). Subjects in the simeprevir group with genotype 1a (without the Q80K baseline polymorphism) and genotype1b HCV demonstrated similar SVR12 rates. A number of demographic and baseline characteristics have been shown to predict a lower SVR rate with interferon-based treatment. These include a high viral load at baseline, advanced disease on liver histology (bridging fibrosis and cirrhosis), older age, African American race, and absence of the IL28B CC genetic polymorphism. Each of these factors impacted efficacy results in both the simeprevir and Control groups in the pivotal phase 3 studies, as anticipated. Most striking in the subgroup analysis was the substantial impact of the Q80K baseline HCV GT1a polymorphism on the efficacy of simeprevir. In subjects with the Q80K polymorphism at baseline, no statistically significant difference in SVR12 rates was observed when comparing the simeprevir group to the Control group. In the pooled C208 and C216 trials, the SVR12 rate in GT1a subjects with the Q80K polymorphism was 58% in the simeprevir group and 55% in the Control Group. In HPC3007, the SVR12 rate in GT1a subjects with the Q80K polymorphism was 47% in the simeprevir group and 30% in the Control Group. A statistically significant difference was appreciated when comparing the SVR12 rates in simeprevir treated GT1a subjects without the Q80K polymorphism to placebo treated subjects without the Q80K polymorphism in both the pooled treatment-naïve studies and in the relapser study (SVR rates of 84% versus 43% and 78% versus 24%, respectively). In all other subgroup analyses presented in Table 2, SVR12 rates were significantly higher in the simeprevir group compared to the Control group.
Replicon culture studies indicated that Q80K expression was associated with an approximately 10-fold reduction in susceptibility to simeprevir relative to wild-type controls, providing mechanistic support for the reduced clinical efficacy against Q80K polymorphic variants.
The Q80K polymorphism is a common polymorphism found in GT1a patients in the U.S. population. The Sponsor performed an analysis pooling all subjects from the C205, C206, C208, C216, and HPC3007 trials, and found that of the 298 GT1a subjects in the U.S with sequencing data, 48% had the Q80K polymorphism at baseline. None of the 113 GT1b subjects in the U.S. with sequencing data had the Q80K polymorphism at baseline.
Given the high frequency of the Q80K polymorphism in the U.S. population and its significant impact on rates of SVR12, DAVP is recommending that all GT1a patients be screened for the Q80K polymorphism. Alternative treatment options should be considered for patients found to be infected with this polymorphic variant. Notably, no Q80K-related reductions in efficacy were observed during the pivotal trials of the currently approved NS3/4A protease inhibitors, telaprevir and boceprevir.
Efficacy in the Partial/Null Responder Population:
The Sponsor has requested an indication for treatment of chronic hepatitis C genotype 1 infection in partial and null responders based on data from the Phase 2b trial, C206. In C206, subjects categorized as partial or null responders received 12, 24, or 48 weeks of simeprevir in combination with PR which was administered for 48 weeks in all treatment arms (refer to Section 2.1 for details on the trial design).
The SVR24 results (the primary endpoint for this study) from the C206 trial by treatment arm and population (including the overall ITT population, prior relapsers, prior partial responders, and prior null responders) are presented in Table 3.
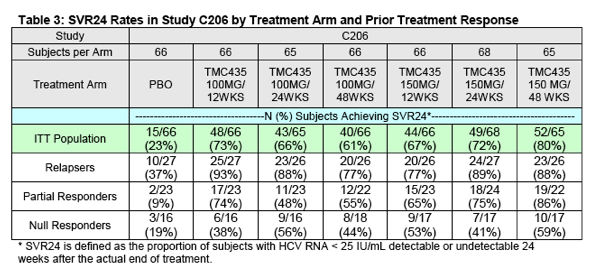
DAVP believes it is reasonable to combine the following two study arms for a pooled efficacy analysis: 1) simeprevir 100 mg for 12 weeks with PR for 48 weeks and 2) simeprevir 150 mg for 12 weeks with PR for 48 weeks. For both of these arms the duration of simeprevir was 12 weeks (the Sponsor's proposed duration for approval). Although the doses in the two arms differ (i.e. 100 mg versus 150 mg), there are no data or scientific reasons to anticipate that the 100 mg dose of simeprevir would prove more effective than the 150 mg dose. Therefore, this could be considered a conservative pooling. The SVR24 data from this pooled analysis are presented in Table 4 below.
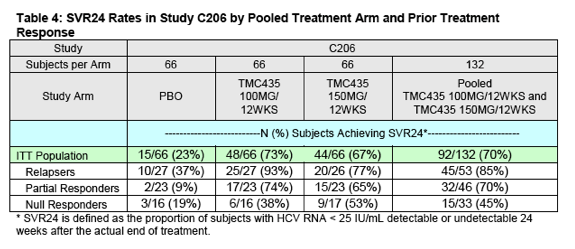
In partial responders, the difference in SVR24 rates between the pooled simeprevir groups and placebo group reached statistical significance (P-value
< 0.0001). In null responders, the difference in SVR24 rates (26%) between the pooled simeprevir groups and placebo group (45% vs. 19%, respectively) did not reach statistical significance (P-value = 0.11). The lack of statistical significance in the null responder population may relate to the small sample size of the groups and greater than predicted SVR24 rates in the null responder placebo group (which was more than twice that of the SVR24 rate in the partial responder placebo group).
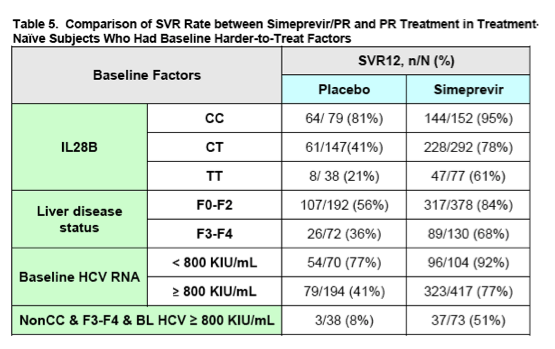
Additional indirect evidence for efficacy in the partial and null responder populations may be drawn from an analysis of the more difficult to treat sub-populations found within the Phase 3 treatment-naïve trials, C208 and C216. Specifically, subjects with baseline factors known to impact the effectiveness of HCV treatment, including IL28B genotypes CT and TT, advanced liver fibrosis (e.g., Metavir score F3-F4), and/or high baseline HCV RNA (e.g., baseline HCV RNA ≥ 800,000 IU/mL), achieved significantly higher SVR rates with simeprevir/PR versus placebo/PR (refer to Table 5 for details).
Based on the above analyses, as well as our experience with the approved NS3/4A protease inhibitors, DAVP believes that it is reasonable to extend the indication of simeprevir for the treatment of partial and null responders. Patients in these populations would receive 12 weeks of simeprevir in combination with PR, followed by an additional 36 weeks of PR. DAVP believes screening for the presence of the Q80K polymorphism (as discussed in detail above) will be especially important in these populations in which SVR is already anticipated to be more difficult to achieve. DAVP intends to designate the submission of the final data from the ongoing Phase 3 trial of simeprevir in combination with PR in partial and null responders (HPC3001) a post-marketing commitment.
2.4 Clinical Pharmacology:
Simeprevir is orally bioavailable. It is highly plasma protein-bound (>99.9%) and distributes to the liver. The primary route of simeprevir elimination is hepatobiliary excretion; urinary excretion is negligible. Simeprevir exhibits a greater-than-dose-proportional increase in exposures. This phenomenon appears to be caused by saturation of hepatic uptake (via OATP1B1/3) and metabolism (via CYP3A4) of simeprevir at doses above 100 mg QD in healthy subjects and 75 mg QD in patients with HCV infection.
Because it is a CYP3A4 substrate, simeprevir exposures increase in the presence of moderate or strong CYP3A inhibitors (as observed in clinical drug-drug interaction studies with erythromycin and ritonavir) and decrease in the presence of moderate or strong CYP3A inducers (as observed in clinical drug-drug interaction studies with efavirenz and rifampin); therefore, coadministration of simeprevir with moderate or strong CYP3A inhibitors or inducers is not recommended. Simeprevir inhibits intestinal (but not hepatic) CYP3A, as demonstrated in a drug-drug interaction study with midazolam administered IV or PO. Simeprevir also inhibits OATP1B1/3: exposures of the OATP1B1/3 substrates rosuvastatin and atorvastatin are higher upon coadministration with simeprevir, indicating that a maximum recommended dose for rosuvastatin and atorvastatin is advisable.
Following administration of simeprevir 150 mg QD, mean simeprevir exposures (AUC24) are 2 to 3-fold higher in HCV-infected patients compared to healthy volunteers. This observation appears to be a function of the selected dose (150 mg, at which CYP3A is saturated) as well as lower functional hepatic CYP3A content in patients with chronic HCV infection1,2,3,4,5, which results in slower simeprevir clearance in patients.
HCV-uninfected subjects with moderate or severe hepatic dysfunction (Child-Pugh B or C, respectively) had mean simeprevir AUC24 values that were approximately 2.4- and 5.2-fold higher, respectively, compared to healthy controls. In addition, HCV-infected patients of East Asian ancestry had mean AUC24 values that were approximately 3.4-fold higher than the pooled Phase 3 population, which was approximately 91% Caucasian. The higher exposures observed in Chinese and Japanese subjects in Phase 1 studies compared to Caucasian subjects prompted evaluation of lower simeprevir daily doses in the ongoing Phase 3 development programs in Japan, China, and Korea. Similar to the elevated exposures observed in subjects with HCV infection, the increased exposures observed in subjects with moderate hepatic impairment and subjects with East Asian ancestry are likely a consequence of the smaller liver volume and lower amount of functional CYP3A and/or OATP1B1/3 in these subpopulations6,7 compared to HCV-uninfected and Caucasian subjects, respectively.
Administration of simeprevir 150 mg QD to patients with moderate or severe hepatic impairment or patients with East Asian ancestry may not be advisable for the following reasons:
1. The safety profile of simeprevir exposures anticipated in patients with East Asian ancestry or patients with moderate or severe hepatic impairment following administration of simeprevir 150 mg QD (i.e. average AUC24
values 2 to 5-fold above the Phase 3 mean) has not been well-characterized in the Phase 3 trials;
2. A positive correlation exists between simeprevir exposures and adverse events, including rash and photosensitivity, as indicated by the exposure-response relationships for safety;
3. No additional therapeutic benefit is gained from higher exposures, as indicated in the Phase 3 studies by the flat exposure-response relationship for efficacy at a simeprevir dose of 150 mg QD;
4. With respect to patients with East Asian ancestry, lower doses of simeprevir (in addition to 150 mg, in some countries) are currently being evaluated in Phase 3 trials in East Asian countries; safety data from these ongoing trials have not been evaluated by DAVP.
Based on the above reasons, DAVP recommends that the dose of simeprevir be reduced for patients with moderate or severe hepatic impairment or patients with East Asian ancestry. However, as no reduced dose strengths are currently available, definitive dose recommendations and labeling for these populations will likely need to be accomplished as postmarketing requirements or commitments.
2.5 Non-Clinical Safety:
The major target organs identified in the simeprevir nonclinical studies include the gastrointestinal tract (vacuolation of apical enterocytes, dilatation of lacteals) and the liver (hepatocellular necrosis, centrilobular hypertrophy, increases in ALT, AST, ALP, and bilirubin).
Simeprevir will be categorized as a Pregnancy Category C in labeling based on reproductive toxicity effects in the pregnant rat and mouse (early mortality and post-implantation loss), the fetus (skeletal variations, adverse body weight decrease) as well as the developing rat offspring (adverse body weight decrease, small size, motor activity decreases). The reproductive effects were seen in the rat at approximately 0.2 to 1 times the mean clinical AUC and in the mouse at 4 times the mean clinical AUC. The potential reproductive toxicity risks will be mitigated by appropriate labeling. Simeprevir is indicated for use in combination with pegylated interferon alfa and ribavirin. Ribavirin has a boxed warning and is contraindicated for use in pregnancy due to potential teratogenic and embryocidal effects. Therefore, the potential risk of simeprevir exposure in pregnant women is low because administration would be avoided during pregnancy due to the indicated use with ribavirin.
|
|
| |
| |
|
|
|