| |
Persistent immune activation in chronic HIV infection:
do any interventions work? (cure research)
|
| |
| |
Download the PDF here
AIDS: POST ACCEPTANCE, 16 January 2013
Rajasuriar, Reena; Khoury, Gabriela; Kamarulzaman, Adeeba; French, Martyn Andrew; Cameron, Paul Urquhart; Lewin, Sharon Ruth
aCentre of Excellence for Research in AIDS (CERiA), University Malaya, Kuala Lumpur, Malaysia, bDepartment of Pharmacy,
Faculty of Medicine, University Malaya, Kuala Lumpur, Malaysia, cDepartment of Infectious Diseases, Monash University,
Melbourne, Australia, dCentre for Virology, Burnet Institute, Melbourne, Australia, eDepartment of Medicine, Monash University,
Melbourne, Australia, fSchool of Pathology and Laboratory Medicine, University of Western Australia, Perth, Australia,
gDepartment of Clinical Immunology, Royal Perth Hospital and PathWest Laboratory Medicine, Perth, Australia, and hInfectious
Diseases Unit, Alfred Hospital, Melbourne, Australia.
Abstract
Persistent immune activation (IA) is a hallmark of chronic HIV infection. IA has been associated with poor CD4 T-cell recovery, non-AIDS defining illnesses and mortality during combination antiretroviral therapy (cART). Measures of chronic immune activation, namely T-cell activation and more recently monocyte activation and plasma inflammatory and thrombotic biomarkers, have all been shown to remain elevated despite years of suppressive cART. Here we review recent clinical trials and therapeutic approaches targeted to reduce persistent IA in HIV patients and discuss the impact of each of these approaches on clinically relevant end-points.
Introduction
The availability of combination antiretroviral therapy (cART) has led to substantial reduction in morbidity and mortality in HIV-infected patients, however life expectancy remains reduced especially in HIV-infected patients who initiate cARTwith a CD4 T-cell counts <200 cells/ml [1]. Increased immune activation (IA) in patients on long-term suppressive cART [2-4] has been associated with increased mortality [5,6] and both AIDS and non-AIDS defining illnesses [7-10], suggesting that chronic IA may have a potential role in driving increased morbidity and mortality.
Causes of HIV-associated immune
activation
The mechanisms driving systemic IA in chronic HIV infection are multifactorial (reviewed in [11], Fig. 1) and include the translocation of microbial products from the gastro-intestinal tract [12,13], low-level HIV viremia [14,15] and co-infections with other persistent viral pathogens including cytomegalovirus (CMV) and hepatitis C virus (HCV) [16]. A recent study in SIV-infected macaques demonstrated a significant increase in IA and coagulation markers, including D-dimers, following exogenous administration of LPS [17]. The excessive production of interferon-alpha (IFN-a) [18-21] and pro-inflammatory cytokines leading to upregulation of pro-apoptotic molecules [22-25], lymph node fibrosis [26],dysfunction of CD4 T-regulatory cells (T-regs) [27,28] and depletion of CD161++/(mucosalassociated invariant T-cells, MAIT) [29,30] are likely to also contribute.
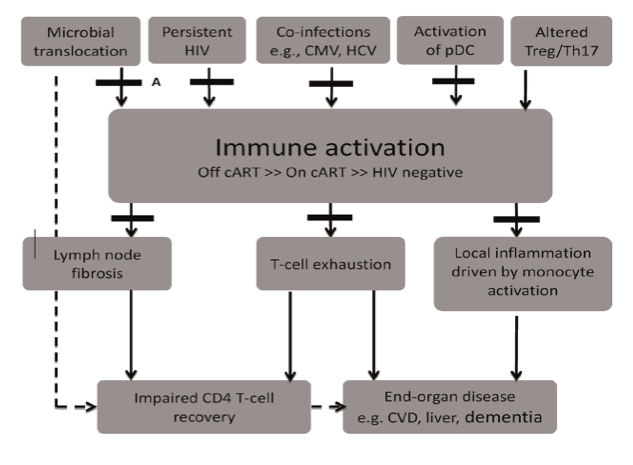
Fig. 1. Schematic representation of the potential causes of chronic immune activation in HIV-infected patients, its impact on clinical end-points and strategies of intervention tested in recent completed and on-going clinical trials. Potential drivers of immune activation include microbial translocation which occurs due to persistent dysfunction in the gut-associated lymphoid tissue (GALT), persistent HIV infection, co-infections with cytomegalovirus (CMV) and hepatitis C, aberrant activation of plasmacytoid dendritic cells (pDCs) and altered ratio of Tregs and Th17 cells. Immune activation (IA), though significantly reduced, persists even in patients receiving suppressive combination antiretroviral therapy (cART) and leads to increased lymphoid tissue fibrosis and T-cell exhaustion which affects CD4 T-cell recovery. Chronic IA also activates monocytes which drives local inflammation in tissues and leads to the development of various end-organ damage and non-AIDS defining illnesses. Various treatment strategies to attenuate immune activation or its effects have recently been trialled and are labelled A to F. These strategies include (A) agents that promote mucosal repair in the GALT (bovine serum colostrum, micronutrient supplementation, pro and pre-biotics); (B) cART treatment intensification (maraviroc and raltegravir); (C) treatment of co-infections (valgancyclovir, interferon-a and ribavirin); (D) agents that reduce pDC activation (chloroquine and hydroxychloroquine); (E) agents that reduce TGF-b1 mediated lymph node fibrosis (pirfenidone); and (F) immune-modulators (HMG CoA reductase inhibitors, minocycline, selective cyclo-oxygenase-2 inhibitors, leflunomide and IVIG). (Figure modified from [113]).
Strategies to reduce persistent immune
activation in HIV-infected individuals
Pharmacological agents
Multiple clinical trials have been completed (Table 1), or are in development (Table 2), to reduce IA in HIV-infected patients and have also been reviewed in [31].
Statins
The use of statins in HIV-infected patients on and off cART have reported variable changes in T-cell activation and highly-sensitive C-reactive protein (hsCRP) levels [32-35] but no effect on CD4? T-cell counts [32,33,36]. However, in two large observational studies of cART treated patients, the use of statins was associated with reduced mortality [37] and reduced incidence of non-Hodgkin lymphoma (NHL) [38]. No immunological correlates were assessed in these two studies and a greater understanding of the mechanisms underlying the benefits of statins is needed.
Chloroquine and hydroxychloroquine
Chloroquine and hydroxychloroquine inhibit endosomal acidification in plasmacytoid dendritic cells (pDCs) and TLR-7 signaling by HIV-1 single stranded (ss)RNA and also inhibit IFN-a production [39,40]. In vitro, chloroquine inhibited pDC activation and maturation, reduced IFN-a mediated CD8 T-cell activation and down-modulated indolamine 2-3 dioxygenase (IDO) and PD-L1 expression on pDCs, which are negative regulators of T-cell responses [41].
A recent RCT in cART-naïve patients (n=13) found chloroquine was associated with decreased memory CD8 T-cell activation, CD4 and CD8 T-cell proliferation, and LPS levels compared to baseline but there were no changes in plasma HIV RNA [42]. In contrast, a RCT of hydroxychloroquine in cART naïve patients demonstrated no change in CD8 and CD4 T-cell activation and proliferation, an increase in HIV RNA and decrease in CD4 T-cell counts [43]. In a small non-randomised study (n=420), administration of hydroxychoroquine to patients receiving suppressive cARTwas associated with a reduction in multiple markers of IA but no significant increase in CD4 T-cell recovery [44]. Given these promising findings, numerous clinical trials are currently being conducted with choloroquine (NCT00819390) and hydroxychloroquine (NCT01232660).
Selective cyclooxygenase-2 (COX-2) inhibitors
Selective COX-2 inhibitors are anti-inflammatory agents which modulate T-cell activation via inhibition of prostaglandin E2 and the cyclic adenosine 30,50-monophosphate (cAMP) protein kinase A pathway ([45,46], reviewed in [47]). In cART-treated patients, selective COX-2 inhibitors were associated with increased T-cell proliferation [48], a non-significant reduction in T-cell activation and increased perforin-containing CD8+ T-cells [49]. A recent RCT of high-dose celecoxib in untreated HIV-infected patients (n1/431) reported a significant reduction in IA levels [50].
Leflunomide
A77 1726, the active metabolite of the anti-rheumatoid arthritis agent leflunomide, has anti-HIV activity [51,52], inhibits pyrimidine synthesis [52,53] and reduces proliferation of activated T-cells in vitro [54]. A small RCT in cART-naïve patients (ALETHIA; n=16), found no significant change in CD4 and CD8 T-cell counts or HIV RNA levels in patients treated with leflunomide compared to placebo [55]. Furthermore, more grade 1 and 2 adverse events were reported with leflunomide. However, short-termleflunomide usewas associated with reduced T-cell cycling and activation. It is currently unclear if similar immunological effects will be seen in patients receiving cART.
Biological agents
Bovine colostrum, micronutrients and pre-/probiotics Multiple approaches are now being taken to directly reduce microbial load and translocation in HIV patients. These include supplementation with micronutrients, bovine colostrum, pro- and prebiotics; all of which have previously been shown to reduce HIV-associated diarrhea [56-60]. These strategies may also alter the composition of gut microflora which may be important in modulating microbial translocation-driven IA [61,62]. Most of these studies have been in cART-naïve patients and some have reported increases in CD4 T-cell counts [59,60,63-67] (Table 1). In a RCTof orally administered hyperimmune bovine colostrum (that contains antibodies to LPS), there was no effect on immune activation or CD4 T-cell recovery in patients receiving suppressive cART [68].
Antiretroviral treatment
Chronic IA in patients receiving cART may also be driven by low-level HIV viremia [14,15,69,70]. In multiple observational and RCT studies, the addition of raltegravir to suppressive cART resulted in no significant IA reduction in plasma, CSF or tissue [68,71-76] nor any change in endothelial function, a surrogate marker of cardiovascular disease [77]. There have however been two studies that have shown that the addition of raltegravir led to a significant reduction in T-cell activation markers in a subset of patients and a reduction in reservoir size [78,79]. Further larger randomised studies are still needed to definitively determine the impact of raltegravir intensification on IA.
Several studies of maraviroc intensification have shown a reduction in IA [80-82] however one study reported an unexpected increase in IA [83] (Table 1). CCR5 antagonists inhibit the binding and signaling of CCR5 ligands (including CCL3, CCL4 and CCL5) leading to an increase in their plasma concentration. This increase could potentially activate monocytes/macrophages via CCR1 [81] and/or increase antigen-specific T-cell and antibody responses which has been observed in some [84] but not all studies [85]. Further studies are needed to better characterize the immunological changes associated with maraviroc use.
The timing of cART initiation may be an important parameter that influences IA. Studies of patients treated during chronic infection have demonstrated persistently elevated IA levels post-cART compared to uninfected controls [2-4]. A recent prospective study of cART initiated during acute infection demonstrated reduced IA to normal levels after 48 weeks [86]. Prospective or randomized trials need to be performed to determine the effect of early versus delayed cARTon IA in patients with chronic infection.
Treatment of co-infections
Anti-CMV treatment-Valgancyclovir
Increased CMV-specific antibodies and/or T-cells have been associated with atherosclerosis [87,88] and impaired CD4 T-cell reconstitution [89] in HIV-infected patients on cART, suggesting that CMV co-infection may be a driver of persistent IA. A RCT with valgancyclovir in CMV-seropositive cART-treated patients (n1/430) found that both CMV DNA and expression of CD38+HLA DR+ on T-cells declined significantly during valgancyclovir therapy [90]. It is currently unclear whether this approach will translate to clinical benefits and the feasibility of prolonged administration of valgancyclovir may be limited by significant toxicities of the drug.
Anti-HCV treatment-interferon gamma and ribavirin
HCV-specific treatment with interferon-a and ribavirin in HIV/HCV co-infected patients receiving cART has been associated with a significant reduction in markers of T-cell activation [91] and endothelial dysfunction [92] however its impact on clinical end-points is currently unknown.
Other strategies including treatment with intravenous immunoglobulin (IVIG) and minocycline have also been trialed in small studies but have yielded negative results. (see Table 1).
Challenges in designing clinical trials to
reduce IA
There aremultiple challenges in designing clinical trials to reduce chronic IA in patients receiving suppressive cART. First, these studies will require patients who are otherwise clinically well to take an additional drug(s) that may be associated with toxicities. Therefore, the risks and benefits need to be carefully assessed. Second, there are multiple markers of IA and inflammation that have been studied and it is currently unclear which best predicts AIDS and non-AIDS related morbidities in patients receiving suppressive cART. Biomarkers such as IL-6, D-dimer and sCD14 show promise as they are relatively easy to standardize from measurements in plasma but whether they are indeed robust markers for predicting clinical outcomes following specific interventions needs further evaluation. Finally, given that clinical events are rare in patients on suppressive cART, relatively large samples sizes will be required to demonstrate a clinically relevant impact of any intervention to reduce IA.
Conclusion
To date most studies aimed at reducing IA have only included a small number of patients and/or shown an effect on biomarkers of IA and have not had the power to assess any effects on clinical outcomes. Given that there are several candidate approaches that have shown promise in small proof of concept trials, these compounds warrant evaluation in larger randomized clinical trials that systematically evaluate both IA biomarkers and clinical outcomes.
| |
| |
| |
|
|
|