| |
Can HIV Be Cured?
|
| |
| |
Download the PDF here
Download the PDF here
from Jules: Here are 2 articles published in the journal Cell, one by the Siliciano group finding that some latent HIV may be not inducible and this first article is a commentary. Both pdfs are attached.
David Margolis says in response to the findingregarding lsrger than expected & more cumbersome barriers & challenges to purging the latent reservoirs:
"I have to say that I am not so bothered by it. Potent, selective drugs could purge virus from millions or even billions of cells in a day --- the challenge will be to do this safely and effectively. The great importance of this work is that it clarifies the understanding of the strengths and weaknesses of our current latency assays."
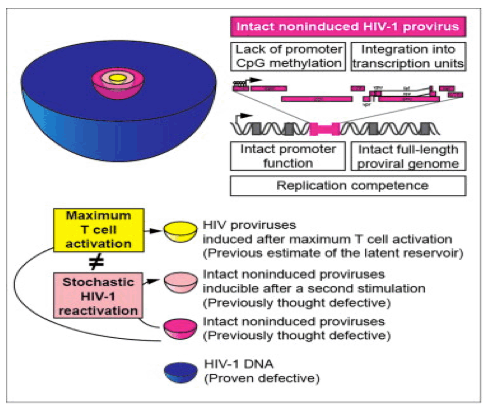
Is All the latent virus inducible?: "We next determined the locations of noninduced proviruses in the host genome to understand whether they were integrated into sites unfavorable for transcription. Bushman et al. showed that HIV-1 typically integrates into transcription units (as shown in Schroder et al., 2002). However, in some model systems, integration into regions of heterochromatin is associated with latency (Jordan et al., 2003). Using inverse PCR at limiting dilution, we found that 92.9% of 70 noninduced proviruses resided in transcription units (Figure 5A), consistent with previous observations in patient resting CD4+ T cells (91%) (Han et al., 2004). Based on transcript levels measured in a serial analysis of gene expression (SAGE) library from a primary cell model of latency (Shan et al., 2011), most noninduced proviruses were integrated into genes transcribed at moderate levels in both resting and activated CD4+ T cells (Figure 5B). Noninduced proviruses were found in both orientations with respect to the host genes (Figure 5C). Overall, these results indicate that noninduced proviruses are not integrated into chromosomal regions that are repressive for transcription; thus, other factors must have prevented expression."
"To determine whether intact noninduced proviruses are permanently silenced or potentially inducible under certain conditions, we tested whether repeated PHA stimulation could induce additional noninduced proviruses (Figure S5). We stimulated multiple replicate cultures of 2 x 105 patient resting CD4+ T cells with PHA in VOA conditions. We then split each culture well equally into two wells on day 7. As all patient cells have divided by day 7 (Figure S1), each split well contained daughter cells derived from cells activated in the original well. One set of the "split-culture" wells was activated again with PHA, while the other set was cultured without additional stimulation. We then compared supernatant p24 levels after another 14 days of culture. Among 126 p24- wells from four patients, 31 (24.6%) became p24+ after the additional round of PHA stimulation, while the paired-culture well that did not receive an additional round of stimulation remained p24- (Figure 7D). It is not yet clear what fraction of the intact noninduced proviruses are inducible in vivo. Nevertheless, these results demonstrate that at least some intact noninduced proviruses can be induced under repeated stimulation."
Study authors say in the Discussion section: "Understanding why intact noninduced proviruses did not produce infectious virus after maximum in vitro T cell activation is critical for determining their clinical significance. Possible explanations include silencing by repressive chromatin modifications or transcriptional interference. We analyzed CpG methylation of the LTRs of noninduced proviruses at the clonal level. In contrast to some in vitro models of HIV-1 latency (Kauder et al., 2009), we found that in patient CD4+ T cells there was little CpG methylation at the LTR, consistent with another recent study (Blazkova et al., 2012). We also examined whether noninduced proviruses are silenced by integration into heterochromatin. We found that most of the noninduced proviruses were integrated into active transcription units, consistent with previous studies showing that most HIV-1 proviruses are integrated into introns of actively transcribed genes in cell lines (Schroder et al., 2002) and patient resting CD4+ T cells (Han et al., 2004). Another potential explanation for the noninduced proviruses is transcriptional interference (Han et al., 2008 and Lenasi et al., 2008). Since T cell activation may overcome transcriptional interference due to the high affinity of NF-κB for its binding sites in the LTR (Lenasi et al., 2008), transcriptional interference may not be a major cause of silencing of the noninduced proviruses.......
........We propose that, despite maximum T cell activation, the induction of latent proviruses is stochastic. Cellular gene expression levels may follow a digital or analog distribution after T cell receptor activation, as a result of stochastic and dynamic processes (Chakraborty and Das, 2010). Elegant experimental and theoretical studies have shown that HIV-1 proviruses may show stochastic fluctuations in expression depending on levels of Tat (Burnett et al., 2009, Singh et al., 2010, Weinberger et al., 2005 and Weinberger et al., 2008). We propose that intact proviruses undergo stochastic induction even after maximum cellular activation. Some will be induced by one round of activation, while others will remain silent but retain the potential to be activated subsequently. These results indicate an increased barrier to cure, as all intact noninduced proviruses need to be eradicated. Underestimation of intact proviruses by VOAs could be reflected in delayed viral rebound after an apparent "cure," and overestimation of LR size resulting from detection of defective proviruses by PCR assays could result in prolonged, excessive exposure to toxic latency reversing agents. Thus, the molecular analysis of noninduced proviruses contributes in an important way to HIV-1 eradication efforts."
Underestimate of HIV reservoirs threatens purging approach
http://www.natap.org/2013/HIV/052613_01.htm
Revealing the Reservoir, and A Second Cure? Comments by Ron Swanstrom
http://www.natap.org/2013/CROI/croi_115.htm
"Comments in Report for NATAP by David Margolis: Ya-Chi Ho from the Siliciano laboratory then presented details of the "non-induced provirus" study introduced by Siliciano in his plenary talk (abstr. 43). As discussed above, while 1 out of 1 million resting CD4+ T cells can be induced to release replication-competent virus, 100 to 1000 out of 1 million resting CD4+ T cells carry HIV-1 proviral DNA. To examine the intermediate step between proviral DNA and produced virions, HIV RNA, Ho amplified cell-associated HIV RNA from tissue culture wells in which replicating viral particles were not detected. These "non-induced proviruses" were then reconstructed by PCR with HIV LTR primers, and internal gag PCR performed to verify clonality (ie there was only one viral RNA species per well). The viral genome was then reconstructed by internal PCR, and sequencing to examine the viral genome performed. Ho found that there were large internal deletions in the genomes of 50% for the RNAs found, deletions in the virion packaging signal in 4%, 10% had internal mutations, and 30% bore C-to-U mutations characteristic of the effect of the human ApoBEC defense system. This left about 12% of the RNA genomes potentially intact. These precise numbers should be considered preliminary, as the work is clearly ongoing, but they should not change very much with further work.
Importantly, Ho then tested the ability of these reconstructed genomes to replicate in PBMCs in culture. Most of these genomes that were identified as "non-induced proviruses" could grow when they were reconstructed and grown in optimal conditions in culture. The proportion of these viruses that might be thought of as an unaccounted-for threat that was quoted here was about 80%, but again this is ongoing work. Nevertheless, this work makes the point that most of the DNA we detect is junk, and the gold-standard outgrowth assay must under-represent the true in vivo frequency of the replication-competent latent viral reservoir. The extent of this under-estimate was said to be up to 50-fold. So at worst, the frequency of latent virus is 50 infected cells per million. While this might sound like terrible news to some, I have to say that I am not so bothered by it. Potent, selective drugs could purge virus from millions or even billions of cells in a day --- the challenge will be to do this safely and effectively. The great importance of this work is that it clarifies the understanding of the strengths and weaknesses of our current latency assays."
------------------------
Stochastic Fate Selection in HIV-Infected Patients
Cell 24 October 2013
Ariel D. Weinberger1 and Leor S. Weinberger2,3,*
1Department of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138, USA
2Virology and Immunology, Gladstone Institutes, San Francisco, CA 94158, USA
3Department of Biochemistry and Biophysics, University of California San Francisco, San Francisco, CA 94158, USA
"By cementing stochasticity as a driver of HIV latency, this study may force a reevaluation of clinical attempts to purge the latent reservoir. First, it appears that the size of the latent reservoir has been underestimated, perhaps substantially. This is because previous studies assumed that, upon cellular activation, any noninduced viruses would be defective rather than latent. More critically, in presuming that cellular activation induces all latent viruses, many thought that the latent population could be deterministically purged. Touting an intervention known as "shock and kill," the idea was to first activate ("shock") patient cells to induce all latent virus. Standard antiretroviral therapy would then purge ("kill") the reactivated viruses, leaving a patient HIV-free. Unfortunately, it now appears that even the most potent "shocks" only reactivate a subset of the latent viruses. Perhaps repeated shocking will be more effective, but each repetition might just be another stochastic roll of the dice. Some virus will likely always emerge latent."
Summary
Classic studies proposed that stochastic variability ("noise") can drive biological fate switching, enhancing evolutionary success. Now, Ho et al. report that HIV's reactivation from dormant (latently infected) patient cells-the major barrier to an HIV cure-is inherently stochastic. Eradicating an incompletely inducible (probabilistic) viral phenotype will require inventive approaches.
From tiny viruses to complex vertebrates, biological systems share a common challenge to preserve reproductive fitness in unpredictable, changing environments. Faced with environmental variability, many organisms evolve complex sensor-actuators to continually gauge their surroundings and deterministically adapt. But, 50 years ago, Dan Cohen proposed an alternate solution: if organisms could stochastically generate a range of phenotypes in each environment, they could "hedge their bets" in much the same way that financial houses diversify their assets to minimize risk against economic crashes. In desert annuals, where reproductive success is governed by unpredictable weather patterns, Cohen noted that fitness could be enhanced if chance governed each seed's fate to germinate or hibernate (e.g., when the husk thickness of each seed is allowed to stochastically vary). With some seeds randomly entering dormancy whatever the environment, the annuals are always left with a long-lived subpopulation to avoid extinction during unforeseen droughts. But what molecular mechanism would allow organisms to probabilistically generate the needed cell-to-cell variability? Years later, studies of active-versus-dormant infection (i.e., lysis-lysogeny) in the bacterial virus phage λ suggested an answer: noisy gene expression (Arkin et al., 1998). Gene expression is, in fact, inherently stochastic, subject to random fluctuations in regulating enzymes, mRNAs, and other biomolecules. These diffusion-driven molecular fluctuations are unavoidable at the single-cell level and appear sufficient to shift cells between transcriptional on and off states (Raj and van Oudenaarden, 2008). With some cells randomly active and others dormant, the result is a distribution of cell fates across a population. A similar distribution of cell fates may now have been found in HIV patients in the clinic. In this issue of Cell, Ho and colleagues (below full text) (Ho et al., 2013 report that HIV's reactivation from lifelong dormant (i.e., "latent") reservoirs is stochastic, likely interfering with persistent therapeutic efforts to activate and purge this problematic reservoir.
The theory that stochastic noise is sufficient to drive cell-fate decisions has been demonstrated in a range of biological systems, from bacteria to vertebrates (Balazsi et al., 2011). Nevertheless, the theory has faced stiff challenge. Alternative hypotheses have argued that hidden deterministic variables, for example the state of the host cell during viral infection or the number of infecting viruses, have a larger impact on eventual cell-fate (St-Pierre and Endy, 2008,Zeng et al., 2010). Unknown and unmeasured, these variables might strongly differ between the disparate cellular phenotypes, in fact predicting the seeming stochasticity. Ever finer and more expansive measurements, it appeared, would be needed to rule out deterministic explanations.
Unexpectedly, a new chapter may now come from a clinical angle. Much like bacteriophage λ, when HIV infects a cell, two outcomes are possible. After HIV integrates its proviral DNA into the genome of CD4+ T cells, either it enters a state of active replication killing the cell or it enters a long-lived latent state where transcription from the provirus is largely quiescent (Finzi et al., 1997). Since these latent cells are unaffected by antiretroviral therapy, which only targets actively replicating HIV, the latent reservoir ensures HIV's lifelong persistence during therapy. Most troublingly, latency is reversible, especially when latently infected cells subsequently activate. As a result, if a patient is removed from therapy, HIV levels quickly resurge from the progeny of reactivated latent viruses, rapidly reaching pretreatment levels (Figure 1A).
With latency being the greatest barrier to curing HIV in patients, the field has continually searched for a set of cellular drivers that might deterministically push HIV into or out of latency. The dominant view has been that latency results from HIV's infrequent infection of "transitioning" T cells, i.e., T cells undergoing the transition from activated to resting-memory states (Figure 1B, left). Insufficiently activated, these cells would not support viral gene expression, due to blocks such as heterochromatin-mediated silencing or a lack of transcriptional activators, for an excellent review see Siliciano and Greene, 2011). Yet, evidence had also been found implicating HIV's own noisy transcriptional positive-feedback circuit in the latency decision (Weinberger et al., 2005). Stochastic depletions in a critical HIV molecule (Tat protein) can prevent active HIV transcription, resulting in a chance of latent infections whatever the target-cell state (Figure 1B, right).
Ho and colleagues did not set out to resolve the stochastic-versus-deterministic argument. As discoverers of HIV's latent reservoir over 15 years ago (Finzi et al., 1997), the Siliciano group sought to better calculate the latent reservoir's size. The standard assay for quantifying the reservoir isolates memory CD4+ T cells from a patient and activates these cells to reactivate latent virus. By supplying the reactivating latent viruses with abundant target cells, infections can be scored and the number of latently infected cells back-calculated. However, not all integrated HIV is induced by cellular activation. Previously, these noninduced viruses were assumed to be defective, a result of HIV's high mutation rate. But, when Ho et al. sequenced these noninduced viruses, 12% of the genomes showed no obvious deletions or inactivating mutations. Moreover, when they synthesized the noninduced intact proviruses, the viruses grew with wild-type kinetics. They went further, analyzing many of the usual suspects of HIV latency, including: viral promoter function, promoter methylation, and viral integration into transcriptionally silent regions. None of these deterministic factors could explain why some viruses were noninduced while seemingly identical viruses were easily reactivated.
As a final test to determine if noninduced proviruses were in fact viable-since maximal cellular induction did not activate these viruses-Ho et al. performed beautiful repeated-stimulation experiments on the patient cells. By all measures, the first stimulation activated 100% of the cells from resting to activated. If latency were a deterministic epiphenomenon governed by cellular activation, or noninduced viruses were nonviable, subsequent stimulations on a completely activated target-cell population would have had little effect on the noninduced virus. Instead, during the second set of stimulation experiments, a subset of patient samples showed significantly more viruses reactivating from latency. While it is always possible that "hidden" deterministic factors were missed, the fact that the same inputs generated distinct outputs is a hallmark of stochasticity.
By cementing stochasticity as a driver of HIV latency, this study may force a reevaluation of clinical attempts to purge the latent reservoir. First, it appears that the size of the latent reservoir has been underestimated, perhaps substantially. This is because previous studies assumed that, upon cellular activation, any noninduced viruses would be defective rather than latent. More critically, in presuming that cellular activation induces all latent viruses, many thought that the latent population could be deterministically purged. Touting an intervention known as "shock and kill," the idea was to first activate ("shock") patient cells to induce all latent virus. Standard antiretroviral therapy would then purge ("kill") the reactivated viruses, leaving a patient HIV-free. Unfortunately, it now appears that even the most potent "shocks" only reactivate a subset of the latent viruses. Perhaps repeated shocking will be more effective, but each repetition might just be another stochastic roll of the dice. Some virus will likely always emerge latent.
For basic virology, these findings raise the striking possibility that stochastic latency evolved to provide retroviruses like HIV with a bet-hedging fitness advantage. This would represent a paradigm shift in retrovirology where latency is currently viewed as a host-driven epiphenomenon with no evolutionary role in the natural history of infection. Viewing latency as an advantageous evolutionary fate decision-much as bacterial persistence and phage lysogeny are viewed-might explain why HIV Tat expression is exceptionally noisy, so noisy that Tat fluctuations alone are sufficient to drive a latency decision in nontransitioning cells (Weinberger et al., 2005). Given HIV's extremely rapid evolution, this noise would likely have been filtered out over the millions of years of natural lentiviral infections were it not selectively beneficial. But, how would noise be advantageous to lentiviruses? Stochastic latency would only provide a bet-hedging fitness advantage if lentiviruses needed to minimize their risks of extinction due to environmental catastrophes. In reality, lentiviruses mutate rapidly enough to evade immune clearance, generate extremely high viral loads, and only infect a small percentage (1%-2%) of environmental target cells. There appears to be little danger of lentiviral population crashes (and lentiviruses clearly did not evolve under pressure from antiretroviral drugs). If latency is, in fact, a viral-mediated stochastic fate decision, one wonders what selection pressures drive its persistence. Future work may address this.
--------------
Replication-Competent Noninduced Proviruses in the Latent Reservoir Increase Barrier to HIV-1 Cure
Cell 24 October 2013
Ya-Chi Ho,1 Liang Shan,1,5 Nina N. Hosmane,1 Jeffrey Wang,2 Sarah B. Laskey,1 Daniel I.S. Rosenbloom,3 Jun Lai,1
Joel N. Blankson,1 Janet D. Siliciano,1 and Robert F. Siliciano1,4,*
1Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA
2Health Sciences Center, School of Medicine, Louisiana State University, New Orleans, LA 70112, USA
3Program for Evolutionary Dynamics, Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA 02138, USA
4Howard Hughes Medical Institute, Baltimore, MD 21205, USA
5Present address: Department of Immunobiology, School of Medicine, Yale University, New Haven, CT 06510, USA
HIGHLIGHTS
A total of 1.7% of noninduced HIV-1 proviruses have intact genomes and LTR function
Reconstructed intact noninduced proviruses are replication competent
They are integrated into transcription units and have no CpG methylation in the LTR
The size of the latent reservoir for HIV-1 may be underestimated by ~60-fold
Summary
Antiretroviral therapy fails to cure HIV-1 infection because latent proviruses persist in resting CD4+ T cells. T cell activation reverses latency, but <1% of proviruses are induced to release infectious virus after maximum in vitro activation. The noninduced proviruses are generally considered defective but have not been characterized. Analysis of 213 noninduced proviral clones from treated patients showed 88.3% with identifiable defects but 11.7% with intact genomes and normal long terminal repeat (LTR) function. Using direct sequencing and genome synthesis, we reconstructed full-length intact noninduced proviral clones and demonstrated growth kinetics comparable to reconstructed induced proviruses from the same patients. Noninduced proviruses have unmethylated promoters and are integrated into active transcription units. Thus, it cannot be excluded that they may become activated in vivo. The identification of replication-competent noninduced proviruses indicates that the size of the latent reservoir-and, hence, the barrier to cure-may be up to 60-fold greater than previously estimated.
"......Some will be induced by one round of activation, while others will remain silent but retain the potential to be activated subsequently. These results indicate an increased barrier to cure, as all intact noninduced proviruses need to be eradicated. Underestimation of intact proviruses by VOAs could be reflected in delayed viral rebound after an apparent "cure," and overestimation of LR size resulting from detection of defective proviruses by PCR assays could result in prolonged, excessive exposure to toxic latency reversing agents. Thus, the molecular analysis of noninduced proviruses contributes in an important way to HIV-1 eradication efforts."
Introduction
Despite prolonged antiretroviral therapy (ART), HIV-1 persists as transcriptionally inactive proviruses in resting memory CD4+ T cells (Chun et al., 1997, Finzi et al., 1997 and Wong et al., 1997). This latent reservoir (LR) has a long half-life, preventing cure by ART alone (Finzi et al., 1997, Siliciano et al., 2003 and Strain et al., 2003). In resting CD4+ T cells, the lack of active forms of key cellular transcription factors (Bohnlein et al., 1988, Duh et al., 1989, Ganesh et al., 2003, Kinoshita et al., 1997, Nabel and Baltimore, 1987 and West et al., 2001) and of HIV-1 Tat and its cellular cofactors (Cujec et al., 1997, Herrmann and Rice, 1995, Jones and Peterlin, 1994, Kao et al., 1987, Selby and Peterlin, 1990 and Tyagi et al., 2010) limits the initiation and elongation, respectively, of viral transcription (Lassen et al., 2004 and Williams and Greene, 2007). The LR may thus be established when activated CD4+ T cells become infected as they revert back to a resting memory state. In addition, DNA methylation and repressive histone modifications may silence proviruses (Blazkova et al., 2009, Coull et al., 2000, He and Margolis, 2002, Kauder et al., 2009, Van Lint et al., 1996, Verdin et al., 1993 and Williams et al., 2006).
A major approach to eradicating HIV-1 involves reversing latency in patients on ART (Richman et al., 2009 and Deeks, 2012). Cells harboring induced proviruses could then be lysed by HIV-1-specific cytolytic T lymphocytes (CTL) (Shan et al., 2012), while new rounds of infection are blocked by ART. Clinical trials exploring this strategy have used the histone deacetylase inhibitors (Lehrman et al., 2005, Archin et al., 2009, Archin et al., 2012 and Contreras et al., 2009).
Accurate measurement of the LR is essential for evaluating eradication strategies. If the LR is eradicated, ART can be discontinued without rebound viremia. Interruption before complete eradication will likely result in rebound (Davey et al., 1999) and repopulation of the LR.
The standard assay for LR size is a viral outgrowth assay (VOA) (Finzi et al., 1997 and Siliciano and Siliciano, 2005) measuring the frequency of resting CD4+ T cells that produce infectious virus after a single round of maximum in vitro T cell activation. Limiting dilutions of resting CD4+T cells are stimulated with the mitogen phytohemagglutinin (PHA), which reverses latency by inducing T cell activation. Released viruses are expanded by addition of CD4+ T lymphoblasts from HIV-1-negative donors. Culture supernatants are examined for exponential viral growth by ELISA for HIV-1 p24. With this assay, the mean frequency of latently infected cells in patients on ART is ~1/106 resting CD4+ T cells (Eriksson et al., 2013).
It has been assumed that LR size can be assessed with agents like PHA that induce uniform T cell activation (Patel et al., 1988 and Hermankova et al., 2003). However, the frequency of latently infected cells detected in the VOA is 300-fold lower than the frequency of resting CD4+ T cells that harbor proviruses detectable by PCR (Eriksson et al., 2013). Thus, at limiting dilution in the VOA, negative wells contain many proviruses, which we designate noninduced proviruses. The noninduced proviruses are generally considered defective but have not been molecularly characterized. The magnitude of the challenge presented by the LR depends on whether noninduced proviruses can be induced in vivo. We present here a molecular characterization of noninduced proviruses.
Discussion
The LR in resting CD4+ T cells is the major barrier to HIV-1 eradication, and as efforts to cure the infection proceed, accurate measurement of LR size will be essential. This study provides a molecular basis for understanding measures of the LR. Through an analysis of proviruses that did not give rise to infectious virus following a single round of T cell activation (noninduced proviruses), we have provided a definitive explanation for the large discrepancy between results of PCR and culture assays of LR size. In addition, the identification of intact noninduced proviruses indicates that the size of the LR may be much greater than previously thought. We reconstructed full-length, intact noninduced proviruses from multiple patients, and all showed growth kinetics comparable to induced proviruses from the same patient and a reference isolate. These intact noninduced proviruses are not detected in standard culture assays but may nevertheless prevent cure. Thus, the present study provides insights into the extent of the challenge posed by the LR and may lead to novel strategies that target intact noninduced proviruses.
We show here that most noninduced proviruses were rendered defective during reverse transcription by APOBEC3G-induced hypermutation (Yu et al., 2004), by internal deletions caused by copy choice recombination during reverse transcription (Sanchez et al., 1997), or by frame-shift or nonsense mutations caused by the error-prone reverse transcriptase (Bebenek et al., 1989). The resulting defective viral genomes can still integrate because only defects at the ends of the genome affect integration. The defective genomes will be detected in most PCR-based assays of proviral DNA, provided that the primer binding sites are intact. Many of the defective proviruses have large internal deletions encompassing the Tat and Rev ORFs and the RRE. Tat-mediated transactivation is required for effective transcriptional elongation (Kao et al., 1987), and the production of virus particles requires that singly spliced and unspliced HIV-1 mRNAs be exported from the nucleus in a Rev-dependent fashion (Malim et al., 1989). Thus, these deleted proviruses may not produce viral proteins, even after successful induction of transcription. The same is true for hypermutated proviruses, which have stop codons in every ORF. Of note, eradication strategies depend on the production of viral proteins, which allows recognition of the infected cells by HIV-1 specific CTL (Shan et al., 2012). Defective proviruses with large internal deletions and/or APOBEC3G-induced hypermutation may not be eliminated even by strategies that effectively eliminate cells carrying replication-competent virus. These considerations highlight the difficulty of assessing eradication strategies with PCR-based assays.
Although difficult and time consuming, the VOA (Eriksson et al., 2013 and Finzi et al., 1997), which has recently been simplified (Laird et al., 2013), does allow detection of cells harboring replication-competent virus. However, the identification of intact, noninduced proviruses raises the possibility that this assay may dramatically underestimate LR size. Several lines of evidence suggest that this is not simply an issue of assay sensitivity. We showed that the PHA stimulation activates all resting CD4+ T cells as assessed by proliferation and activation marker expression. Using prolonged culture and sensitive RT-PCR assays, we also verified that wells from which noninduced proviruses were obtained were truly negative for viral outgrowth. It is also unlikely that these p24- wells remained negative because of reduced viral fitness, as we showed that intact noninduced proviruses had growth kinetics comparable to induced viruses from p24+ wells. Taken together, these results confirm that we are examining a population of intact proviruses that were not induced to produce infectious virus after a single round of maximum in vitro activation.
To prove replication competence, we reconstructed six intact, noninduced proviruses from six different p24- wells from four patients. Surprisingly, all reconstructed viruses replicated as well as the standard reference isolate and control viruses reconstructed from p24+ wells. A sterilizing cure requires elimination of all replication-competent HIV-1; therefore, the discovery that intact noninduced proviruses are replication competent means that the number of proviruses that must be eliminated is much higher than previously thought. We conservatively estimate that the number may be ~60-fold higher than estimates based on the VOA. Some statistical models suggest an even higher number (medians of 97-273-fold). Of note, there is large interpatient variation in this and other measures of LR size. Overall, our results indicate that the "shock and kill" strategy (Archin et al., 2012 and Deeks, 2012) is challenged with a large but unmeasured hidden population of replication-competent proviruses. It is interesting that, despite the intense search for novel latency reversing agents, none of the drugs tested to date reaches the robust level of in vitro HIV-1 induction achieved by PHA. Thus, the finding that the true size of the LR may be ~60-fold greater than that estimated using PHA activation is particularly disturbing. However, it is also important to point out that even a low level of virus gene expression may be sufficient to allow the elimination of infected cells by an appropriately primed CTL response (Shan et al., 2012) and that the critical variable may be the fraction of latently infected cells induced to express HIV-1 genes.
Understanding why intact noninduced proviruses did not produce infectious virus after maximum in vitro T cell activation is critical for determining their clinical significance. Possible explanations include silencing by repressive chromatin modifications or transcriptional interference. We analyzed CpG methylation of the LTRs of noninduced proviruses at the clonal level. In contrast to some in vitro models of HIV-1 latency (Kauder et al., 2009), we found that in patient CD4+ T cells there was little CpG methylation at the LTR, consistent with another recent study (Blazkova et al., 2012). We also examined whether noninduced proviruses are silenced by integration into heterochromatin. We found that most of the noninduced proviruses were integrated into active transcription units, consistent with previous studies showing that most HIV-1 proviruses are integrated into introns of actively transcribed genes in cell lines (Schroder et al., 2002) and patient resting CD4+ T cells (Han et al., 2004). Another potential explanation for the noninduced proviruses is transcriptional interference (Han et al., 2008 and Lenasi et al., 2008). Since T cell activation may overcome transcriptional interference due to the high affinity of NF-κB for its binding sites in the LTR (Lenasi et al., 2008), transcriptional interference may not be a major cause of silencing of the noninduced proviruses.
We propose that, despite maximum T cell activation, the induction of latent proviruses is stochastic. Cellular gene expression levels may follow a digital or analog distribution after T cell receptor activation, as a result of stochastic and dynamic processes (Chakraborty and Das, 2010). Elegant experimental and theoretical studies have shown that HIV-1 proviruses may show stochastic fluctuations in expression depending on levels of Tat (Burnett et al., 2009, Singh et al., 2010, Weinberger et al., 2005 and Weinberger et al., 2008). We propose that intact proviruses undergo stochastic induction even after maximum cellular activation. Some will be induced by one round of activation, while others will remain silent but retain the potential to be activated subsequently. These results indicate an increased barrier to cure, as all intact noninduced proviruses need to be eradicated. Underestimation of intact proviruses by VOAs could be reflected in delayed viral rebound after an apparent "cure," and overestimation of LR size resulting from detection of defective proviruses by PCR assays could result in prolonged, excessive exposure to toxic latency reversing agents. Thus, the molecular analysis of noninduced proviruses contributes in an important way to HIV-1 eradication efforts.
Results
Transwell VOA Achieves Maximum In Vitro Activation and Outgrowth
To analyze proviruses that did not give rise to infectious virus in the VOA (noninduced proviruses), we first established that the conditions were sufficient to activate 100% of resting CD4+ T cells. Resting CD4+ T cells from patients on suppressive ART for >6 months were labeled with carboxyfluorescein succinimidyl ester (CFSE) and stimulated with PHA and irradiated allogeneic peripheral blood mononuclear cells (PBMC) under conditions used in the VOA. By day 7, >99.8% of patient cells had divided at least once (Figure S1A available online), confirming that PHA causes uniform T cell activation.
In the VOA, viruses released after reversal of latency replicate in healthy donor CD4+ lymphoblasts added to the cultures. To facilitate isolation of noninduced proviruses, we tested whether comparable levels of activation and viral outgrowth could be achieved in transwell cultures in which patient cells were separated from donor lymphoblasts by a cell-impermeable membrane (Figure S1B). In side-by-side comparison with standard VOA cultures from ten patients, transwell cultures showed comparable cellular activation in both p24+ and p24- wells, as >95% of patient cells expressed human leukocyte antigen-DR (HLA-DR) and/or CD25 on day 21 (Figure S1C). Transwell cultures also showed viral outgrowth comparable to standard VOA cultures (Figure S1D). Noninduced proviruses were thus isolated from p24- wells of limiting dilution transwell and standard cultures.
Clonal Amplification and Sequencing of Noninduced Proviruses
We obtained near full-length clonal sequences of noninduced proviruses from eight patients on suppressive ART. Patient characteristics are in Table S1. Noninduced proviruses were obtained from wells seeded with 4 x 104 or 2 x 105 resting CD4+ T cells that were p24- on day 21. In clonal VOA cultures, wells with replicating virus are p24+ by days 10-14 (Laird et al., 2013). Even with a more sensitive real-time (RT)-PCR assay for HIV-1 mRNA (Laird et al., 2013), none of the p24- wells showed exponential growth. Thus, the noninduced proviruses were obtained from wells with no replicating virus, despite maximal T cell activation.
Noninduced proviruses were amplified in limiting dilution PCRs to avoid in vitro recombination. A near-full-length 9.1 kb outer PCR (Li et al., 2007) spanning U5 to U5 (positions 623-9,686, HXB2 coordinates) was followed by nested inner PCRs (Table S2). Aliquots from outer PCRs were first amplified in a nested inner gag PCR (Figure 1A). Cell dilutions for which <20% of the inner reactions were positive were selected because positive wells at these dilutions have >90% probability of being clonal. Aliquots from the gag+ clonal outer PCRs were amplified using four sets of inner PCR primers to obtain fragments overlapping by 150-3,173 base pairs (bp) (Figure 1A). Of note, instead of cloning PCR products, we directly sequenced them. This dramatically reduces PCR errors, because errors occurring after the first or second cycle are present in too small a fraction of the final products to be observed. Sequences with double peaks or nonidentical overlap regions were discarded. We identified 213 noninduced proviruses from eight patients.
Hypermutation and Large Internal Deletions Render Most Noninduced Proviruses Defective
Most (88.3%) noninduced proviruses had obvious defects precluding replication (Figure 1C). Direct sequencing of the nested gag PCR product revealed that ~1/3 (32.4%) of noninduced proviruses had APOBEC3G-mediated G -> A hypermutation occurring in the expected sequence context (GG or GGG) (Yu et al., 2004).
Hypermutated proviruses are replication defective due to start codon mutations and numerous tryptophan -> stop codon mutations (Figure S2). Although the gag gene was analyzed here, other regions of the genome show even greater hypermutation (Yu et al., 2004). Of note, it is unlikely that hypermutated proviruses could produce functional viral proteins due to stop codons in most open reading frames (ORFs).
Noninduced proviruses that were not hypermutated were further analyzed by nested amplification of four overlapping subgenomic fragments (Figure 1A). Of the 144 clonal noninduced proviruses without hypermutation, 97 had large internal deletions identified by smaller amplicon size in electrophoresis (Figure 1B). We mapped the deletion junctions in 58 of these clones (Figure 2A). For example, clone 10CB7_48H1 (Figure 1B) gave a smaller amplicon for the nested C reaction, and amplification of fragments A, B, and D failed due to deletion of nucleotides 4,869-9,533. All 58 mapped deletions would affect expression of the essential regulatory proteins Tat and Rev (Figure 2A) because the deletions encompass the tat and rev exons, the splice sites, and the Rev-responsive element (RRE).
Deletions are not unexpected. HIV-1 is prone to recombination due to pseudodiploidity (two RNA copies per virion with physical proximity for recombination). Frequent template switching events occur during reverse transcription (Simon-Loriere and Holmes, 2011). Switching between short repeats in a single-genome results in deletion of the intervening sequence and one repeat (Temin, 1993). Large deletions have been observed in unfractionated PBMC from viremic patients (Sanchez et al., 1997). Several lines of evidence suggest that these deletions occur in vivo rather than during in vitro analysis. First, deletions were observed following direct sequencing of uncloned PCR products. Second, for a given provirus, the same deletion junctions were observed in different nested PCRs using different primers. Third, short amplicons were not seen in control experiments with plasmids carrying the reference proviral genomes NL4-3 and BaL. Plasmids were mixed, diluted to eight copies per 105 human genome equivalents, and amplified under the same conditions. No deletion or recombination was observed. Fourth, short sequence repeats were identified at some deletion junctions (Figure 2B), consistent with a single polymerase jump due to copy choice recombination during reverse transcription of the minus strand (Sanchez et al., 1997). Taken together, these results demonstrate that a large fraction of noninduced proviruses are nonfunctional due to large internal deletions, likely introduced during reverse transcription.
The precise fraction of proviruses with deletions could be underestimated by this analysis because deletions could affect PCR primer binding sites. For 39 clones, the exact deletion junction could not be identified, probably because the deletions encompassed binding sites for primers used in the nested PCRs. For these clones, we obtained at least two sequences to ensure that the amplicons contained nonhypermutated, patient-specific sequences. For 12 mapped deletions, the deletion included the reverse gag primer binding site, but mapping was possible because other nested reactions were successful.
Mutations in cis Elements Render Some Noninduced Proviruses Defective
Proviruses with correct amplicon size were directly sequenced. A small fraction (8/213) had nonsense mutations and/or frame-shifting insertions or deletions in one or more ORFs. Small deletions (8-98 bp) were found in the packaging signal (χ) in 12 sequences (Figure S3A). The deletions encompassed the major splice donor (MSD) site. Point mutations in the MSD site were found in two proviruses. Full genome sequencing showed that seven of the noninduced proviruses with χ or MSD mutations were otherwise intact. To determine whether these mutations rendered noninduced proviruses defective, we reconstructed three clones by genome synthesis as described later. The reconstructed proviruses included two clones with short (8 bp and 16 bp) χ deletions in packaging stem loop 2 and one with a MSD site mutation (TG|GT -> TG|GG). Although these clones had intact ORFs, they did not replicate in healthy donor CD4+ lymphoblasts (Figure S3B) under conditions in which other reconstructed proviruses replicated well (discussed later). Thus, mutations in cis elements render otherwise intact proviruses defective.
A Significant Proportion of Noninduced Proviruses Are Replication Competent
Of 213 noninduced proviruses, 25 (11.7%) had intact ORFs and cis elements. When compared with induced proviruses from the same patient, no known lethal mutations were seen. To determine whether these intact noninduced proviruses are replication competent, we used the direct sequencing results to reconstruct full-length noninduced proviral clones by de novo genome synthesis (Figure 3A). This strategy avoids PCR and cloning-induced errors. We reconstructed six noninduced proviruses from four patients by inserting the synthesized sequence into a plasmid carrying the reference isolate NL4-3. Not captured in our PCR strategy is a 108 bp segment of the provirus, representing nucleotides (nt) 565-672 (HXB2 coordinates). This segment in the 5' untranslated region includes part of U5 and the primer binding site (pbs) (Figure 3A). Although U5 deletions may not affect replication competence (Vicenzi et al., 1994), we took additional steps to make the reconstructed clones fully patient derived, without any NL4-3 sequence. We used limiting dilution PCR to amplify the LTR-gag region from cells in p24- wells. Using a 424 bp segment from the 5' U3-R-U5 region (HXB2 nt 140-564), we constructed phylogenetic trees (Figure S4). Then, using site-directed mutagenesis, we corrected the 108 bp segment from NL4-3 to the phylogenetically closest sequence from the same patient (Figure 3A). This process results in proviruses that are 100% patient derived and 98.2% equivalent to specific proviruses present in vivo, with the remaining 1.8% equivalent to a very closely related provirus from the same patient. Given the high sequence conservation in the 108 bp segment (Figure S4), we estimated that the reconstructed clones could differ from the parent clones by, at most, 3 nt, or 0.03% of the genome. For each patient, we also reconstructed an induced viral clone from a p24+ well.
It is striking that all six reconstructed noninduced proviruses from four different patients showed replication fitness comparable to that of the NL4-3 and reconstructed induced proviruses from the same patients (Figure 3B). It is unlikely that all of these intact noninduced proviruses could have actually been defective with inactivating mutations in the 108 bp segment that was not directly sequenced, as we showed that an additional round of PHA stimulation causes some noninduced proviruses to produce replication-competent virus (described later). Taken together, these results indicate that a substantial fraction of noninduced proviruses are intact and capable of generating infectious virus if induced in vivo.
Noninduced Proviruses Have Intact Promoter Function unless Hypermutated
The ability of intact noninduced proviruses to produce infectious virus suggests that, at the primary sequence level, their promoters are functional. To confirm this, we cloned long terminal repeats (LTRs) from representative noninduced proviruses into a luciferase reporter construct (Yang et al., 2009). We measured luciferase activity in transfected resting CD4+ T cells before and 4 hr after stimulation with phorbol myristate acetate (PMA) and ionomycin. We also analyzed LTRs from induced proviruses from the same patients and NL4-3. In general, LTRs from noninduced proviruses showed basal (Figures 4A and 4C) and stimulated (Figures 4B and 4D) activity comparable to LTRs from induced proviruses and NL4-3. Decreased LTR function was only observed for hypermutated clones. This likely reflects G -> A hypermutation in binding sites for the transcription factors NF-κB and Sp1 (Figure 4E). Thus, most noninduced proviruses have LTRs that are intact at the primary sequence level.
Most Noninduced Proviruses Are Integrated into Active Transcription Units
We next determined the locations of noninduced proviruses in the host genome to understand whether they were integrated into sites unfavorable for transcription. Bushman et al. showed that HIV-1 typically integrates into transcription units (as shown in Schroder et al., 2002). However, in some model systems, integration into regions of heterochromatin is associated with latency (Jordan et al., 2003). Using inverse PCR at limiting dilution, we found that 92.9% of 70 noninduced proviruses resided in transcription units (Figure 5A), consistent with previous observations in patient resting CD4+ T cells (91%) (Han et al., 2004). Based on transcript levels measured in a serial analysis of gene expression (SAGE) library from a primary cell model of latency (Shan et al., 2011), most noninduced proviruses were integrated into genes transcribed at moderate levels in both resting and activated CD4+ T cells (Figure 5B). Noninduced proviruses were found in both orientations with respect to the host genes (Figure 5C). Overall, these results indicate that noninduced proviruses are not integrated into chromosomal regions that are repressive for transcription; thus, other factors must have prevented expression.
Lack of CpG Methylation in the LTR of Noninduced Proviruses
We next examined whether noninduced proviruses were silenced by CpG methylation. CpG islands are present in the HIV-1 genome (Chavez et al., 2011), including one in the U3 region of the LTR, which contains critical transcription factor binding sites (Figure 6). Therefore, DNA from freshly isolated resting CD4+ T cells and from cells in p24- wells was treated with bisulfite, and then the LTR region was amplified under limiting dilution conditions (Blazkova et al., 2012). Direct sequencing was used to determine the extent of CpG methylation. Only 3.1% of the LTR CpGs were methylated in resting CD4+ T cells from study patients. Even fewer (0.9%) of the CpGs in the LTRs of noninduced proviruses were methylated (Figure 6). In contrast, we readily detected methylation at a CpG island in the env region (75.5%), indicating that this method did not selectively amplify nonmethylated sites. Although CpG methylation at the LTR clearly is well documented in some models of HIV-1 latency (Kauder et al., 2009), our results indicate that noninduced proviruses are not silenced through CpG methylation at the 5' LTR.
Intact Noninduced Proviruses May Increase LR Size by ~60-Fold
The aforementioned results indicate that although most noninduced proviruses have identifiable lethal defects, a substantial fraction are intact and replication competent at the primary sequence level. Analysis of LTR function, integration sites, and methylation status suggests that these intact noninduced proviruses could be induced in vivo, thereby increasing LR size. We compared the frequency of induced proviruses (defined using the VOA) and intact noninduced proviruses (quantitated as the product of total proviral DNA frequency and the fraction of noninduced proviruses that are intact) among the total pool of proviruses (measured by quantitative PCR). Bayesian analysis was chosen instead of maximum likelihood estimation because the former provides nonzero point estimates for patients from whom no clones with intact genomes were identified.
The positive VOA results in every patient and the successful detection of intact noninduced proviruses in patients for whom >20 clones were analyzed suggest that intact noninduced proviruses could be detected in every patient if enough clones are examined. The fraction of intact noninduced proviruses was calculated as the median of an empirical Bayesian posterior, the most conservative of five models tested (Table S3), with a prior distribution chosen to reflect the observed data. Both the fraction of intact noninduced proviruses and the total number of proviruses per 106 resting CD4+ T cells varied dramatically from patient to patient (Figure 7A). There was no correlation between the VOA and the frequency of intact noninduced proviruses (Figure 7B). All statistical models (Table S3) indicated that the median frequency of intact noninduced proviruses was at least ~60-fold higher than the frequency of induced proviruses detected in the VOA. If the intact noninduced proviruses described here can be induced in vivo, then the size of the LR is much greater than previously thought (Figure 7C).
To determine whether intact noninduced proviruses are permanently silenced or potentially inducible under certain conditions, we tested whether repeated PHA stimulation could induce additional noninduced proviruses (Figure S5). We stimulated multiple replicate cultures of 2 x 105 patient resting CD4+ T cells with PHA in VOA conditions. We then split each culture well equally into two wells on day 7. As all patient cells have divided by day 7 (Figure S1), each split well contained daughter cells derived from cells activated in the original well. One set of the "split-culture" wells was activated again with PHA, while the other set was cultured without additional stimulation. We then compared supernatant p24 levels after another 14 days of culture. Among 126 p24- wells from four patients, 31 (24.6%) became p24+ after the additional round of PHA stimulation, while the paired-culture well that did not receive an additional round of stimulation remained p24- (Figure 7D). It is not yet clear what fraction of the intact noninduced proviruses are inducible in vivo. Nevertheless, these results demonstrate that at least some intact noninduced proviruses can be induced under repeated stimulation.
|
|
| |
| |
|
|
|