| |
Dolutegravir plus Abacavir-Lamivudine vs Efavirenz/TDF/FTC Once Daily for the Treatment of HIV-1 Infection
|
| |
| |
Download the PDF here
N Engl J Med Nov 6 2013
-------------------------------
Safety and Efficacy of Dolutegravir (DTG; GSK1349572) in Treatment-Experienced HIV-1 Infected Adolescents: 24-week Results from IMPAACT P1093 - (10/04/13)
Activity of Dolutegravir (DTG) 50 mg BID vs Placebo (PBO) Over 7 Days of Functional Monotherapy in Patients Harbouring Raltegravir- and/or Elvitegravir-Resistant Virus: Primary Endpoint Results of the VIKING-4 Study (ING116529) - (10/16/13)
Phase 3 Assessment of Dolutegravir (DTG) 50 mg Twice Daily (BID) in HIV-1-Infected Subjects With Raltegravir (RAL) and/or Elvitegravir (EVG) Resistance in VIKING-3: Week 24 Results of All 183 Subjects Enrolled......http://www.natap.org/2013/IAS/IAS_21.htm
48 Week Bone Marker Changes in Dolutegravir (DTG; GSK1349572) Plus Abacavir/Lamivudine (ABC/3TC) vs Tenofovir/Emtricitabine/Efavirenz (EFV/TDF/FTC): The SINGLE Trial - (09/13/13)
ID Week: Low Risk but Twice Higher Risk of Suicidal Thoughts or Acts on First-Line Efavirenz - - (10/07/13)
Dolutegravir in Key Demographic Subgroups of Treatment-Experienced HIV-Infected Populations - (09/16/13)
----------------------------
Dolutegravir plus Abacavir-Lamivudine vs Efavirenz/TDF/FTC Once Daily for the Treatment of HIV-1 Infection
N Engl J Med Nov 6 2013
Sharon L. Walmsley, M.D., Antonio Antela, M.D., Ph.D., Nathan Clumeck, M.D.,
Dan Duiculescu, M.D., Andrea Eberhard, M.D., Felix GutiŽrrez, M.D.,
Laurent Hocqueloux, M.D., Franco Maggiolo, M.D., Uriel Sandkovsky, M.D.,
Catherine Granier, D.E.S.S., Keith Pappa, Pharm.D., Brian Wynne, M.D.,
Sherene Min, M.D., and Garrett Nichols, M.D., for the SINGLE Investigators*
"At week 48, the proportion of participants with an HIV-1 RNA level of less than 50 copies per milliliter was significantly higher in the DTG-ABC-3TC group than in the EFV-TDF-FTC group (88% vs. 81%, P=0.003), thus meeting the criterion for superiority.......We designed Study ING114467 (SINGLE) to assess the safety and efficacy of dolutegravir at a dose of 50 mg plus a fixed-dose combination of abacavir-lamivudine, as compared with fixed-dose efavirenz-tenofovir DF-emtricitabine, which is the only single-tablet regimen currently preferred in the U.S. HIV treatment guidelines1,2 and one of two currently recommended single-tablet regimens in the European treatment guidelines.3........The proportion of participants who discontinued therapy owing to adverse events was lower in the DTG-ABC-3TC group than in the EFV-TDF-FTC group (2% vs. 10%); rash and neuropsychiatric events (including abnormal dreams, anxiety, dizziness, and somnolence) were significantly more common in the EFV-TDF-FTC group, whereas insomnia was reported more frequently in the DTG-ABC-3TC group. No participants in the DTG-ABC-3TC group had detectable antiviral resistance; one tenofovir DF-associated mutation and four efavirenz-associated mutations were detected in participants with virologic failure in the EFV-TDF-FTC group."
"Resistance did not develop during treatment to any of the regimen components in any of the participants in the DTG-ABC-3TC group - a finding similar to that in a pivotal phase 3 study of dolutegravir versus raltegravir.12 By contrast, in the latter study, resistance to integrase developed in 5% of participants and resistance to NRTI developed in 20% of the participants with protocol-defined virologic failure who received the raltegravir-containing regimen.12 Furthermore, in SINGLE, four participants with virologic failure in the EFV-TDF-FTC group had NNRTI resistance mutations that developed during treatment, and one additional participant in the EFV-TDF-FTC group had a tenofovir DF-associated resistance mutation. The accumulating clinical data from this study and others (along with in vitro tests including viral passage and integrase binding assays) suggest that there may be a higher barrier to resistance with dolutegravir than with other integrase inhibitors.8,12-14,32,33"
"Mild, nonprogressive increases in the serum creatinine level were observed with dolutegravir, as have been described in previous phase 2b and 3 studies.7,9,12,23 In vitro work has identified the likely mechanism as the blockade of creatinine secretion by means of inhibition of the renal transporter organic cation transporter 2, with no effect on the actual glomerular filtration rate as measured by the clearance of iohexol.27 A similar effect has been reported with use of trimethoprim,28 cobicistat,13,27,29 and the NNRTI rilpivirine.30,31 The effect of dolutegravir on the serum creatinine level occurs early and plateaus quickly (within approximately 2 weeks after the initiation of therapy). Given the effect of dolutegravir on creatinine secretion, a small increase in serum creatinine is anticipated when treatment with the drug is initiated; this change is not clinically meaningful, because it does not reflect changes in the actual glomerular filtration rate. Progressive changes in the creatinine level over time (or changes that emerge after the first month of therapy) are likely to be associated with other causes that require investigation (e.g., nephrotoxic drugs, HIV-associated nephropathy, or obstruction)."
Dolutegravir plus Abacavir-Lamivudine for the Treatment of HIV-1 Infection
Background
Dolutegravir (S/GSK1349572), a once-daily, unboosted integrase inhibitor, was recently approved in the United States for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in combination with other antiretroviral agents. Dolutegravir, in combination with abacavir-lamivudine, may provide a simplified regimen.
Methods
We conducted a randomized, double-blind, phase 3 study involving adult participants who had not received previous therapy for HIV-1 infection and who had an HIV-1 RNA level of 1000 copies per milliliter or more. Participants were randomly assigned to dolutegravir at a dose of 50 mg plus abacavir-lamivudine once daily (DTG-ABC-3TC group) or combination therapy with efavirenz-tenofovir disoproxil fumarate (DF)-emtricitabine once daily (EFV-TDF-FTC group). The primary end point was the proportion of participants with an HIV-1 RNA level of less than 50 copies per milliliter at week 48. Secondary end points included the time to viral suppression, the change from baseline in CD4+ T-cell count, safety, and viral resistance.
Results
A total of 833 participants received at least one dose of study drug. At week 48, the proportion of participants with an HIV-1 RNA level of less than 50 copies per milliliter was significantly higher in the DTG-ABC-3TC group than in the EFV-TDF-FTC group (88% vs. 81%, P=0.003), thus meeting the criterion for superiority. The DTG-ABC-3TC group had a shorter median time to viral suppression than did the EFV-TDF-FTC group (28 vs. 84 days, P<0.001), as well as greater increases in CD4+ T-cell count (267 vs. 208 per cubic millimeter, P<0.001). The proportion of participants who discontinued therapy owing to adverse events was lower in the DTG-ABC-3TC group than in the EFV-TDF-FTC group (2% vs. 10%); rash and neuropsychiatric events (including abnormal dreams, anxiety, dizziness, and somnolence) were significantly more common in the EFV-TDF-FTC group, whereas insomnia was reported more frequently in the DTG-ABC-3TC group. No participants in the DTG-ABC-3TC group had detectable antiviral resistance; one tenofovir DF-associated mutation and four efavirenz-associated mutations were detected in participants with virologic failure in the EFV-TDF-FTC group.
Conclusions
Dolutegravir plus abacavir-lamivudine had a better safety profile and was more effective through 48 weeks than the regimen with efavirenz-tenofovir DF-emtricitabine. (Funded by ViiV Healthcare; SINGLE ClinicalTrials.gov number, NCT01263015.)
The initial antiretroviral therapy (ART) recommended in treatment guidelines for human immunodeficiency virus (HIV) type 1 (HIV-1) infection consists of two nucleoside reverse-transcriptase inhibitors (NRTIs) and a third agent: a nonnucleoside reverse-transcriptase inhibitor (NNRTI; e.g., efavirenz), a ritonavir-boosted protease inhibitor (e.g., atazanavir or darunavir), or an integrase inhibitor (e.g., raltegravir). Integrase inhibitors are the most recent drug class to be approved on the basis of their efficacy and safety profiles.1-3 Over the past 15 years, combination products have been developed, consisting initially of partial regimens with two NRTIs and now available in triple combinations.4,5 All the regimens that have been compared with efavirenz-tenofovir disoproxil fumarate (DF)-emtricitabine or its components have been shown to be noninferior.
Dolutegravir is an unboosted integrase inhibitor with a long plasma half-life (approximately 14 hours) that supports once-daily dosing without the need for pharmacokinetic boosting.6 Dolutegravir in combination with abacavir-lamivudine is being developed as a single-tablet regimen for the treatment of HIV infection. Such treatment could offer advantages over currently available single-tablet regimens and regimens containing alternate drug classes, owing to the absence of effects related to the cytochrome P-450 enzyme CYP3A4 (and thus fewer relevant drug interactions), the absence of tenofovir DF (possibly resulting in improved renal or bone safety), and activity against transmitted viruses that are resistant to NNRTIs.7-9 We designed Study ING114467 (SINGLE) to assess the safety and efficacy of dolutegravir at a dose of 50 mg plus a fixed-dose combination of abacavir-lamivudine, as compared with fixed-dose efavirenz-tenofovir DF-emtricitabine, which is the only single-tablet regimen currently preferred in the U.S. HIV treatment guidelines1,2 and one of two currently recommended single-tablet regimens in the European treatment guidelines.3
Methods
Study Oversight
SINGLE is an ongoing, phase 3, randomized, double-blind study involving participants with HIV-1 infection who had not received therapy previously. We enrolled participants in North America, Europe, and Australia between February 1 and June 13, 2011; the last participant completed 48 weeks of treatment on May 14, 2012.
The sponsor, ViiV Healthcare, participated in the design of the study and in the collection, analysis, and interpretation of the data. All the authors had full access to all the study data. All the authors are responsible for the veracity and completeness of the data reported and vouch for the fidelity of the study to the protocol. An author who was an employee of GlaxoSmithKline wrote the first draft of the manuscript, all the authors reviewed drafts of the manuscript, and the first author had the final responsibility for the decision to submit the manuscript for publication.
Approval by the ethics committee was obtained at each participating center in accordance with the principles of the 2008 Declaration of Helsinki and the Good Clinical Practice guidelines of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use.10 All the participants provided written informed consent before any study-specific procedures were performed. An independent data and safety monitoring committee performed five separate reviews of unblinded efficacy and safety data during the course of the study.
Study Design and Participants
Eligible participants were 18 years of age or older, had HIV-1 infection, had not previously received ART, had a plasma HIV-1 RNA level of at least 1000 copies per milliliter without genotypic evidence of viral resistance at screening, and were negative for the HLA-B*5701 allele. Resistance screening at baseline ensured the activity of all components of the study treatments, and HLA-B*5701 screening, as recommended by treatment guidelines,11 minimized the potential of a hypersensitivity reaction to abacavir. Women who were pregnant or breast-feeding, persons with moderate or severe hepatic impairment, and persons with an estimated creatinine clearance of less than 50 ml per minute were excluded from the study (Table S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org).
Randomization was stratified according to the plasma HIV-1 RNA level at the time of screening (˛100,000 copies per milliliter vs. >100,000 copies per milliliter) and the CD4+ T-cell count (˛200 per cubic millimeter vs. >200 per cubic millimeter). Participants were randomly assigned, in a 1:1 ratio, to the DTG-ABC-3TC group, in which participants received dolutegravir at a dose of 50 mg as a separate tablet and abacavir-lamivudine in a fixed-dose combination of 600 mg and 300 mg, respectively (Epzicom or Kivexa, ViiV Healthcare), or to the EFV-TDF-FTC group, in which participants received an efavirenz-tenofovir DF-emtricitabine tablet once daily at fixed doses of 600 mg, 300 mg, and 200 mg, respectively (Atripla, Bristol-Myers Squibb and Gilead Sciences). Randomization was performed in block sizes of six, with the use of a central procedure. In addition, participants in the DTG-ABC-3TC group received a placebo matching the efavirenz-tenofovir DF-emtricitabine tablet and those in the EFV-TDF-FTC group received placebos matching the dolutegravir and abacavir-lamivudine tablets (i.e., all participants received three tablets each day) (Table S2 in the Supplementary Appendix).
Study End Points
The primary efficacy end point was the proportion of participants with a plasma HIV-1 RNA level of less than 50 copies per milliliter at week 48, as determined with the use of the Snapshot algorithm from the Food and Drug Administration (described in the Statistical Analysis section), which is now widely used in HIV trials.12-15 Secondary efficacy end points included the time to viral suppression (i.e., an HIV-1 RNA level of <50 copies per milliliter) and the change from baseline in the CD4+ T-cell count. Other secondary end points included the safety profile, health outcomes, and the incidence of the development of genotypic and phenotypic resistance to dolutegravir, efavirenz, and the respective backbone-therapy components (abacavir-lamivudine and tenofovir DF-emtricitabine) during the treatment period. (For full details of the study design, see the protocol and the statistical analysis plan, available at NEJM.org.)
Assessments
Study visits were scheduled at baseline and at weeks 2, 4, 8, 12, 16, 24, 32, 40, and 48. After the week 48 visit, participants continued to receive blinded treatment until week 96, with visits scheduled every 12 weeks. On completion of the week 96 visit, all the participants were offered the opportunity to continue the treatment until week 144 in an open-label fashion. The Abbott RealTime HIV-1 assay was used to detect the plasma level of HIV-1 RNA (lower limit of detection, 40 copies per milliliter). CD4+ T-cell counts were assessed by means of flow cytometry in a central laboratory. Adverse events, serious adverse events, and laboratory measurements (including hematologic measurements, fasting lipid profile, and blood-chemistry profile) were assessed at each visit and graded according to the criteria of the Division of the Acquired Immunodeficiency Syndrome at the National Institute of Allergy and Infectious Diseases of the National Institutes of Health.16
Viral genotype was analyzed (Quest Diagnostics) at the screening visit. Samples for resistance testing were obtained at baseline and at the time of protocol-defined virologic failure (defined as two consecutive HIV-1 RNA values of at least 50 copies per milliliter on or after week 24). Participants were required to withdraw from the study if protocol-defined virologic failure was confirmed before week 48. Testing for resistance in all participants with protocol-defined virologic failure included determination of HIV-1 reverse-transcriptase and integrase genotype and phenotype with the use of commercial assays (Monogram Biosciences). Participant-reported measures of health outcomes were assessed with the use of the 20-item, self-reported Symptom Distress Module at baseline and at weeks 4, 24, 48, and 96, as described previously.17,18
Statistical Analysis
According to the protocol, we could conclude that treatment with dolutegravir and abacavir-lamivudine was noninferior to treatment with efavirenz-tenofovir DF-emtricitabine if the lower boundary of a two-sided 95% confidence interval for the difference in the primary end point was less than 10 percentage points lower in the DTG-ABC-3TC group than in the EFV-TDF-FTC group. This margin is consistent with that in other trials in this population.19 Assuming a 75% response rate in the EFV-TDF-FTC group, we calculated that 394 participants who could be evaluated would need to be included in each group for the study to have 90% power to determine the noninferiority of dolutegravir and abacavir-lamivudine, at a one-sided significance level of 2.5%. Efficacy and safety analyses were performed in the intention-to-treat population and safety population, respectively; both populations included all participants who underwent randomization and received at least one dose of study drug. The two populations were identical in this study.
A sensitivity analysis of the primary end point was performed in the per-protocol population, which comprised the intention-to-treat population with the exclusion of participants with a protocol deviation that met prespecified criteria (Table S3 in the Supplementary Appendix). Tests of homogeneity were assessed at 10% to ensure that the treatment difference was maintained across the stratification subgroups: for the plasma HIV-1 RNA level at baseline, the P value was 0.83 for the comparison of the treatment difference between participants with a high viral load and those with a low viral load, and for the CD4+ T-cell count at baseline, the P value was 0.41 for the comparison of participants with a high CD4+ T-cell count and those with a low CD4+ T-cell count.
In the Snapshot algorithm (Table S2 and Fig. S1 in the Supplementary Appendix), participants whose last available HIV-1 RNA value in the analysis window (i.e., from week 42 through week 54) was less than 50 copies per milliliter were considered as having had a response; participants whose HIV-1 RNA level was 50 copies per milliliter or higher in the analysis window or who did not have available data in the analysis window were considered as not having had a response.15 Since the background regimen was part of the blinded randomized treatment, no changes in regimen were permitted in this trial. The adjusted difference in the proportions of participants with a response (the proportion in the DTG-ABC-3TC group minus the proportion in the EFV-TDF-FTC group) was based on a stratified analysis that used Cochran-Mantel-Haenszel weights for the HIV-1 RNA level and CD4+ T-cell count at baseline.20
If both the per-protocol and intention-to-treat analyses showed the noninferiority of dolutegravir and abacavir-lamivudine, testing for superiority was to be conducted. In addition, if the intention-to-treat analysis showed noninferiority, the following two superiority comparisons were also prespecified to be tested, with the use of the fallback procedure21,22 to adjust for the risk of a false positive result, at the 1% and 3% a priori levels, respectively: the time to viral suppression (with the use of the generalized Wilcoxon test) and the change from baseline in the CD4+ T-cell count at week 48 (with the use of a repeated-measures model with adjustment for stratification factors).
Results
Participants
Of the 844 participants who underwent randomization, 833 received at least one dose of study medication (Figure 1. Adherence to treatment was similar in the two study groups; 3 participants (2 participants in the DTG-ABC-3TC group and 1 in the EFV-TDF-FTC group) were excluded from the per-protocol population owing to an interruption of the study drug for more than 10% of the total time of treatment (Table S3 in the Supplementary Appendix). Demographic and disease characteristics at baseline were well balanced between the treatment groups.
The median age of the study participants was 35 years; 16% of the participants were women, 24% were black, and 4% were in class C of the Centers for Disease Control and Prevention HIV classification system (defined as the presence of specific opportunistic infections) (Table S4 in the Supplementary Appendix). The median HIV-1 RNA level at baseline was 4.68 log10 copies per milliliter, and the median CD4+ T-cell count was 338 per cubic millimeter. More participants in the EFV-TDF-FTC group than in the DTG-ABC-3TC group withdrew from the trial prematurely (84 and 51 participants, respectively), most commonly owing to adverse events (Figure 1).
Efficacy
A rapid and sustained virologic response was observed, with 88% of the participants in the DTG-ABC-3TC group, as compared with 81% of those in the EFV-TDF-FTC group, having the primary end point of a plasma HIV-1 RNA level of less than 50 copies per milliliter at week 48 (Figure 2A. The adjusted treatment difference between the two groups was 7 percentage points (95% confidence interval [CI], 2 to 12), with dolutegravir and abacavir-lamivudine meeting the noninferiority criterion. In addition, the dolutegravir and abacavir-lamivudine regimen was shown to be statistically superior to the efavirenz-tenofovir DF-emtricitabine regimen (P=0.003). Overall differences in response (intention-to-treat analysis) were due primarily to discontinuations because of adverse events (10 of 414 participants [2%] in the DTG-ABC-3TC group and 42 of 419 [10%] in the EFV-TDF-FTC group) (Table 1.
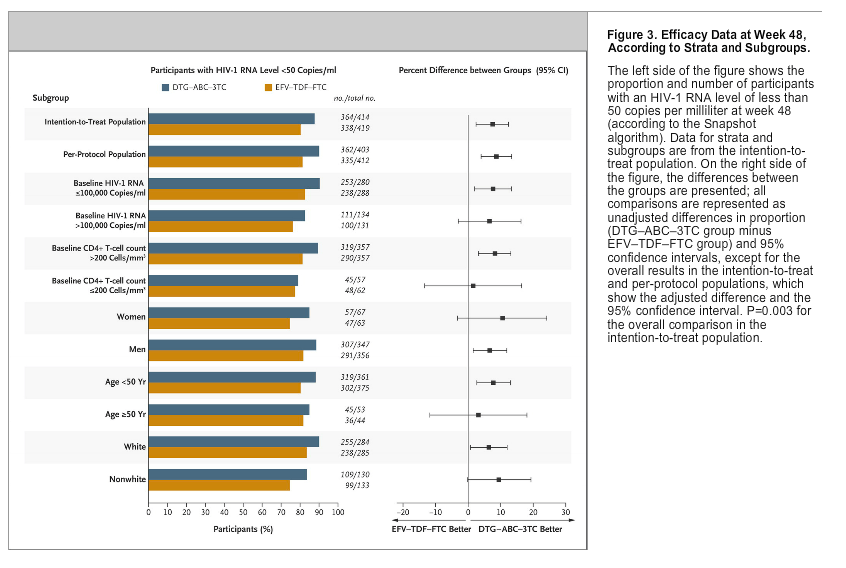
Similar results were observed in the per-protocol population, from which 2% of the participants were excluded (11 of 414 participants [3%] in the DTG-ABC-3TC group and 7 of 419 participants [2%] in the EFV-TDF-FTC group), owing to a number of reasons, including the use of prohibited medication (in <1% of participants). In this analysis, 90% of the participants in the DTG-ABC-3TC group and 81% in the EFV-TDF-FTC group had an HIV-1 RNA level of less than 50 copies per milliliter (Figure 3. The adjusted treatment difference between the two groups was 9 percentage points (95% CI, 4 to 13), again supporting the superiority of the dolutegravir and abacavir-lamivudine regimen. If both the per-protocol and intention-to-treat analyses showed noninferiority, then testing for superiority was to be conducted as described above. No P values were derived for the test of superiority for the per-protocol population; the confidence interval was sufficient to conclude superiority because it excluded 0. Data from the per-protocol population are presented only to support the results in intention-to-treat population.
The difference of 7 percentage points in the treatment response (in the intention-to-treat analysis) in favor of dolutegravir and abacavir-lamivudine was observed among participants with a high baseline HIV-1 RNA level (>100,000 copies per milliliter) and among those with a low baseline HIV-1 RNA level (˛100,000 copies per milliliter); treatment differences were also maintained across key demographic subgroups, including subgroups defined according to race, sex, and age (Figure 3). The median time to viral suppression (HIV-1 RNA level of <50 copies per milliliter) was 28 days among participants receiving dolutegravir and abacavir-lamivudine, as compared with 84 days among those receiving efavirenz-tenofovir DF-emtricitabine (nominal P<0.001) (Fig. S2 in the Supplementary Appendix). The adjusted mean change from baseline at week 48 in the CD4+ T-cell count was greater with dolutegravir and abacavir-lamivudine than with efavirenz-tenofovir DF-emtricitabine (267 per cubic millimeter vs. 208 per cubic millimeter; nominal P<0.001) (Figure 2B). There were no significant changes from baseline in measures of health outcomes in either treatment group.
Safety
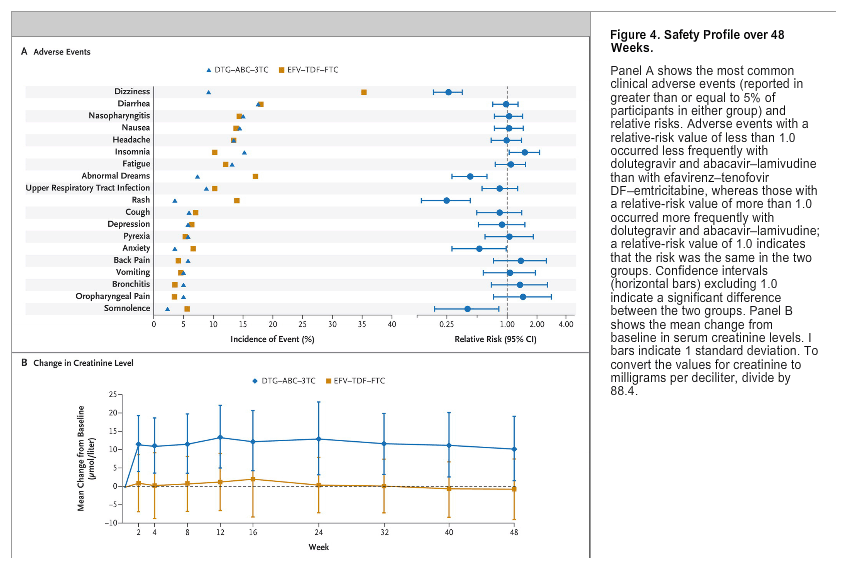
The 48-week safety profile of dolutegravir and abacavir-lamivudine was generally favorable, as compared with that of efavirenz-tenofovir DF-emtricitabine (Figure 4, and Table S5 in the Supplementary Appendix). During the 48-week observation period of this study, diarrhea, nasopharyngitis, nausea, headache, and fatigue were among the most commonly reported clinical adverse events (mainly mild to moderate in severity). Overall, adverse events of grade 3 or 4 were reported in 10% of the participants in the DTG-ABC-3TC group and in 16% of those in the EFV-TDF-FTC group. Rash and neuropsychiatric events (including abnormal dreams, anxiety, dizziness, and somnolence) were significantly more common with efavirenz-tenofovir DF-emtricitabine, whereas insomnia was reported more frequently with dolutegravir and abacavir-lamivudine (Figure 4A). Insomnia events were typically mild in intensity and led to the discontinuation of the study drug in one participant in the DTG-ABC-3TC group and in two in the EFV-TDF-FTC group. No cases of myocardial infarction or other ischemic coronary events were reported through week 48.
Drug-related adverse events (as assessed by the investigator) were reported more frequently among participants who received efavirenz-tenofovir DF-emtricitabine than among those who received dolutegravir and abacavir-lamivudine (66% vs. 43%), as were adverse events leading to discontinuation of the study drug. The most frequent adverse events leading to the discontinuation of efavirenz-tenofovir DF-emtricitabine (i.e., those that occurred in >2% of the participants) were psychiatric and nervous system disorders; in the DTG-ABC-3TC group, adverse events leading to discontinuation of the regimen occurred in two or fewer participants (<1%) in each of the various categories (Table 1).
Overall, nine participants were considered by the investigator to have had a serious adverse event that was related to the study drug: one participant (<1%) in the DTG-ABC-3TC group (with suspected drug hypersensitivity) and eight (2%) in the EFV-TDF-FTC group (four participants with psychiatric events, two with drug hypersensitivity, one with a cerebrovascular accident, and one with renal failure). The incidence of serious adverse events was similar in the two groups (Table S6 in the Supplementary Appendix). Through week 48, two participants, both in the EFV-TDF-FTC group, died; these deaths were considered to be unrelated to the study drug (sepsis with renal failure in one participant and pneumonia in one), although the accompanying renal failure in the one participant with sepsis was considered to be possibly related to the study medication.
Participants receiving dolutegravir and abacavir-lamivudine had small mean increases in the serum creatinine level, a finding consistent with that in previous studies of dolutegravir.12,23 In our study, the mean increases, which ranged from 10.2 to 13.4 μmol per liter (0.12 to 0.15 mg per deciliter), were evident by week 2 and subsequently remained stable through week 48 (Figure 4B). No significant change from baseline was observed in the urinary albumin-to-creatinine ratio (with urinary albumin measured in milligrams per liter and creatinine in micromoles per liter; the ratio value was multiplied by 1000 to achieve a ratio of milligrams per millimoles). The median ratio was 0.00 in the DTG-ABC-3TC group (interquartile range, -0.30 to 0.30) and 0.05 in the EFV-TDF-FTC group (interquartile range, -0.20 to 0.30).
The distribution and number of graded clinical biochemical and hematologic events that developed while the participants were receiving treatment were similar in the two groups. Elevations in alanine aminotransferase, aspartate aminotransferase, bilirubin, and alkaline phosphatase levels of grade 2, 3, or 4 that developed while the participants were receiving treatment occurred in similar proportions in the two groups (Table 1). None of the participants had concurrent increases in alanine aminotransferase and bilirubin levels.
Virology
Through week 48, a total of 4% of the participants in each group met the criteria for protocol-defined virologic failure and had resistance testing performed (Table S7 in the Supplementary Appendix). The majority of these participants had low-level viremia, with 16 of 18 participants who received dolutegravir and abacavir-lamivudine and 11 of 17 who received efavirenz-tenofovir DF-emtricitabine having an HIV-1 RNA level of less than 200 copies per milliliter at the time of virologic failure. Among the participants in the DTG-ABC-3TC group, no major NRTI or integrase-inhibitor resistance mutations were detected with the use of commercial testing. In the EFV-TDF-FTC group, 1 participant had the tenofovir DF-associated resistance mutation K65K/R, and 4 had NNRTI resistance mutations (Table S7 in the Supplementary Appendix).
Discussion
The efficacy of dolutegravir plus abacavir-lamivudine was superior to that of the combination therapy of efavirenz-tenofovir DF-emtricitabine, which is a recommended therapy in guidelines for persons with HIV infection.1-3 The superior responses were driven primarily by a lower rate of discontinuation due to adverse events in the DTG-ABC-3TC group than in the EFV-TDF-FTC group. The treatment differences favoring dolutegravir plus abacavir-lamivudine were similar in participants with HIV-1 RNA levels of up to 100,000 copies per milliliter and in those with HIV-1 RNA levels above 100,000 copies per milliliter and were also maintained across key demographic subgroups, including subgroups defined according to race, sex, and age. Finally, statistically superior responses in favor of dolutegravir plus abacavir-lamivudine were also observed with respect to the change from baseline in CD4+ T-cell counts at week 48.
The response rates at week 48 were consistent with rates reported in several studies involving participants who had not previously received ART in which integrase-inhibitor regimens, efavirenz regimens, or both were used,12,13,24 including one study showing that dolutegravir was noninferior to raltegravir with respect to virologic success (88% and 85% of the participants with a response to treatment [HIV-1 RNA level, <50 copies per millimeter], respectively).12 In the current study, the 81% response rate with efavirenz-tenofovir DF-emtricitabine was similar to the rate seen in two pivotal studies that used this regimen as the comparator.13,24 In the current study, the percentage of participants receiving efavirenz-tenofovir DF-emtricitabine who had adverse events leading to withdrawal (10% of the participants in the group) was less than that observed in a recent cohort study, in which 89 of 472 participants (19%) discontinued efavirenz-tenofovir DF-emtricitabine during the first year of therapy, owing primarily to neuropsychiatric toxic events.25
In this HLA-B*5701-negative population, the safety profile of dolutegravir plus abacavir-lamivudine was generally favorable, as compared with the safety profile of efavirenz-tenofovir DF-emtricitabine. Dolutegravir was associated with significantly lower reported rates of neuropsychiatric and rash events than have been described previously with efavirenz.25,26 No episodes of serious rash (e.g., the Stevens-Johnson syndrome, toxic epidermal necrolysis, and erythema multiforme) were reported in either treatment group. The rate of insomnia (15%) was higher than in previous studies of dolutegravir (5% of the participants in two other phase 3 trials9,12 and 2% of those in a phase 2b trial7,23). SINGLE was the only randomized study of dolutegravir that included a targeted questionnaire, including specific questions on insomnia. Insomnia events were generally mild and led to discontinuation of the study drug in only one participant in the DTG-ABC-3TC group.
Mild, nonprogressive increases in the serum creatinine level were observed with dolutegravir, as have been described in previous phase 2b and 3 studies.7,9,12,23 In vitro work has identified the likely mechanism as the blockade of creatinine secretion by means of inhibition of the renal transporter organic cation transporter 2, with no effect on the actual glomerular filtration rate as measured by the clearance of iohexol.27 A similar effect has been reported with use of trimethoprim,28 cobicistat,13,27,29 and the NNRTI rilpivirine.30,31 The effect of dolutegravir on the serum creatinine level occurs early and plateaus quickly (within approximately 2 weeks after the initiation of therapy). Given the effect of dolutegravir on creatinine secretion, a small increase in serum creatinine is anticipated when treatment with the drug is initiated; this change is not clinically meaningful, because it does not reflect changes in the actual glomerular filtration rate. Progressive changes in the creatinine level over time (or changes that emerge after the first month of therapy) are likely to be associated with other causes that require investigation (e.g., nephrotoxic drugs, HIV-associated nephropathy, or obstruction).
Resistance did not develop during treatment to any of the regimen components in any of the participants in the DTG-ABC-3TC group - a finding similar to that in a pivotal phase 3 study of dolutegravir versus raltegravir.12 By contrast, in the latter study, resistance to integrase developed in 5% of participants and resistance to NRTI developed in 20% of the participants with protocol-defined virologic failure who received the raltegravir-containing regimen.12 Furthermore, in SINGLE, four participants with virologic failure in the EFV-TDF-FTC group had NNRTI resistance mutations that developed during treatment, and one additional participant in the EFV-TDF-FTC group had a tenofovir DF-associated resistance mutation. The accumulating clinical data from this study and others (along with in vitro tests including viral passage and integrase binding assays) suggest that there may be a higher barrier to resistance with dolutegravir than with other integrase inhibitors.8,12-14,32,33
This study has some limitations. First, only 16% of the participants were women, which reflects the difficulties in recruiting women in long-term studies with strict requirements regarding birth control and the exclusion of pregnant women. Second, the proportion of participants with a CD4+ T-cell count of less than 200 per cubic millimeter was relatively low (but reflective of evolving guidelines for starting therapy). Two studies involving participants who had previously received treatment are currently being conducted (SAILING33 and VIKING-3 [ClinicalTrials.gov number, NCT01328041]34); these two ongoing studies will provide data on the effectiveness of dolutegravir in persons with baseline immunodeficiency that is more severe than that in the participants in our study. Long-term follow-up will be useful to further define any differences between treatments with respect to the emergence of resistance as well as the efficacy and safety profiles.
We are continuing to follow participants in the two treatment groups through 144 weeks of therapy. In the SPRING-2 study, the data at 96 weeks showed continued efficacy with no new issues regarding safety or toxic events.35 Monitoring of efficacy and safety in the ongoing SINGLE trial, through more than 96 weeks, has shown similar persistence of efficacy and no new safety signals. The primary results from these studies suggest that dolutegravir may provide people living with HIV infection with an additional initial treatment option.
|
|
| |
| |
|
|
|