 |
|
|
| |
Ledipasvir and Sofosbuvir for Previously Treated HCV Genotype 1 Infection
|
| |
| |
Download the PDF here
Download the PDF here
Download the PDF here
Nezam Afdhal, M.D., K. Rajender Reddy, M.D., David R. Nelson, M.D., Eric Lawitz, M.D., Stuart C. Gordon, M.D., Eugene Schiff, M.D., Ronald Nahass, M.D., Reem Ghalib, M.D., Norman Gitlin, M.D., Robert Herring, M.D., Jacob Lalezari, M.D., Ziad H. Younes, M.D., Paul J. Pockros, M.D., Adrian M. Di Bisceglie, M.D., Sanjeev Arora, M.D., G. Mani Subramanian, M.D., Ph.D., Yanni Zhu, Ph.D., Hadas Dvory-Sobol, Ph.D., Jenny C. Yang, Pharm.D., Phillip S. Pang, M.D., Ph.D., William T. Symonds, Pharm.D., John G. McHutchison, M.D., Andrew J. Muir, M.D., Mark Sulkowski, M.D., and Paul Kwo, M.D. for the ION-2 Investigators
Background
Effective treatment for hepatitis C virus (HCV) genotype 1 infection in patients who have not had a sustained virologic response to prior interferon-based therapy represents an unmet medical need.
Methods
We conducted a phase 3, randomized, open-label study involving patients infected with HCV genotype 1 who had not had a sustained virologic response after treatment with peginterferon and ribavirin, with or without a protease inhibitor. Patients were randomly assigned to receive the NS5A inhibitor ledipasvir and the nucleotide polymerase inhibitor sofosbuvir in a once-daily, fixed-dose combination tablet for 12 weeks, ledipasvir-sofosbuvir plus ribavirin for 12 weeks, ledipasvir-sofosbuvir for 24 weeks, or ledipasvir-sofosbuvir plus ribavirin for 24 weeks. The primary end point was a sustained virologic response at 12 weeks after the end of therapy.
Results
Among the 440 patients who underwent randomization and were treated, 20% had cirrhosis and 79% had HCV genotype 1a infection. The rates of sustained virologic response were high in all treatment groups: 94% (95% confidence interval [CI], 87 to 97) in the group that received 12 weeks of ledipasvir-sofosbuvir; 96% (95% CI, 91 to 99) in the group that received 12 weeks of ledipasvir-sofosbuvir and ribavirin; 99% (95% CI, 95 to 100) in the group that received 24 weeks of ledipasvir-sofosbuvir; and 99% (95% CI, 95 to 100) in the group that received 24 weeks of ledipasvir-sofosbuvir and ribavirin. No patient discontinued treatment owing to an adverse event. The most common adverse events were fatigue, headache, and nausea.
Conclusions
Treatment with a once-daily, single-tablet regimen of ledipasvir and sofosbuvir resulted in high rates of sustained virologic response among patients with HCV genotype 1 infection who had not had a sustained virologic response to prior interferon-based treatment. (Funded by Gilead Sciences; ION-2 ClinicalTrials.gov number, NCT01768286.)
Among the estimated 170 million people in the world who have chronic hepatitis C virus (HCV) infection, approximately 60% have the genotype 1 strain of the virus.1 The treatment of patients infected with HCV genotype 1 is evolving rapidly.2-6 At the end of 2013, the Food and Drug Administration (FDA) approved two new direct-acting antiviral agents for the treatment of HCV infection: the nucleotide polymerase inhibitor sofosbuvir (Gilead Sciences) and the protease inhibitor simeprevir (Janssen Therapeutics).7,8
Among the regimens that have been approved by the FDA for patients with HCV genotype 1 infection who have not had a sustained virologic response to prior interferon-based therapy - historically, the population hardest to cure - are 12 weeks of sofosbuvir with peginterferon and ribavirin or 24 to 48 weeks of simeprevir with peginterferon and ribavirin. The only interferon-free option currently approved for HCV genotype 1 infection is 24 weeks of sofosbuvir and ribavirin for patients who are ineligible to receive interferon because of absolute or relative contraindications. A guideline recently issued by the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America recommends 12 weeks of sofosbuvir and simeprevir, with or without ribavirin, for previously treated patients with HCV genotype 1 infection (on the basis of limited data from phase 2 trials).9,10
Ledipasvir (Gilead Sciences) is a new HCV NS5A inhibitor with potent antiviral activity against HCV genotypes 1a and 1b.11 In phase 2 trials, the combination of ledipasvir and sofosbuvir, with and without ribavirin, resulted in high rates of sustained virologic response among patients with HCV genotype 1 infection who had received prior treatment with interferon-based regimens, including patients who had received a protease-inhibitor regimen and those with compensated cirrhosis.12,13 To simplify the regimen and improve adherence to therapy, ledipasvir and sofosbuvir have been combined in a single fixed-dose tablet for use once daily (ledipasvir-sofosbuvir). We conducted a phase 3 trial of 12 or 24 weeks of treatment with ledipasvir-sofosbuvir, with and without ribavirin (Ribasphere, Kadmon Pharmaceuticals), in patients with HCV genotype 1 infection who did not have a sustained virologic response after treatment with either peginterferon, ribavirin, and a protease inhibitor or peginterferon and ribavirin; the trial included patients with cirrhosis.
Methods
Patients
From January 3, 2013, to February 26, 2013, at 64 sites in the United States, we enrolled patients 18 years of age or older who had chronic HCV genotype 1 infection. Eligible patients were those who had not had a sustained virologic response with either peginterferon and ribavirin or an NS3/4A protease inhibitor combined with peginterferon and ribavirin. Patients who had discontinued prior treatment owing to an adverse event were not eligible. All the patients provided written informed consent.
Approximately 20% of the enrolled patients had evidence of cirrhosis, defined by a liver-biopsy specimen showing evidence of cirrhosis (Metavir stage 4 [on a scale from 0 to 4, with higher stages indicating a greater degree of fibrosis] or Ishak score of 5 or 6 [on a scale from 0 to 6, with higher scores indicating a greater degree of fibrosis]) or a FibroTest score of more than 0.75 (on a scale of 0 to 1, with higher scores indicating more severe fibrosis) and an aspartate aminotransferase:platelet ratio index of more than 2 (with higher scores indicating a greater likelihood of extensive fibrosis). There were no upper limits on age or body-mass index. All the eligibility criteria are listed in the study protocol (available with the full text of this article at NEJM.org).
Study Design
In this randomized, open-label trial, all the patients received a fixed-dose combination tablet containing 90 mg of ledipasvir and 400 mg of sofosbuvir, administered orally once daily. Ribavirin was administered orally twice daily, with the dose determined according to body weight (1000 mg daily in patients with a body weight of <75 kg, and 1200 mg daily in patients with a body weight ≥75 kg).
Patients were randomly assigned in a 1:1:1:1 ratio to one of four treatment groups: ledipasvir-sofosbuvir for 12 weeks, ledipasvir-sofosbuvir plus ribavirin for 12 weeks, ledipasvir-sofosbuvir for 24 weeks, or ledipasvir-sofosbuvir plus ribavirin for 24 weeks. Randomization was stratified according to genotype (1a vs. 1b), presence or absence of cirrhosis, and response to prior therapy (relapse or virologic breakthrough vs. no response). See the Supplementary Appendix, available at NEJM.org, for definitions of all types of response to prior therapy.
Study Oversight
This study was approved by the institutional review board or independent ethics committee at each participating site and was conducted in compliance with the principles of the Declaration of Helsinki, Good Clinical Practice guidelines, and local regulatory requirements. The study was designed and conducted according to the protocol by the sponsor (Gilead Sciences) in collaboration with the academic investigators. The sponsor collected the data, monitored the study conduct, and performed the statistical analyses. An independent data and safety monitoring committee reviewed the progress of the study.
The investigators, participating institutions, and sponsor agreed to maintain confidentiality of the data. All the authors had access to the data and assume responsibility for the integrity and completeness of the data and analyses reported. The first draft of the manuscript was prepared by a professional writer who is an employee of Gilead Sciences, an author who is also an employee of Gilead Sciences, and the first author, with input from all the authors.
Study Assessments
In addition to laboratory and clinical tests, the screening assessments included serum HCV RNA genotyping, measurement of the HCV RNA level, and IL28B genotyping. The serum HCV RNA level was measured with the COBAS TaqMan HCV Test, version 2.0, for use with the High Pure System (Roche Molecular Systems), which has a lower limit of quantification of 25 IU per milliliter. See the Supplementary Appendix for more information on study assessments.
End Points
The primary efficacy end point was the rate of sustained virologic response, defined as the absence of quantifiable HCV RNA in serum (<25 IU per milliliter), at 12 weeks after the end of therapy among all patients who underwent randomization and were treated. Secondary end points included the rate of sustained virologic response at 24 weeks after the end of treatment.
Statistical Analysis
The primary statistical hypotheses of the study were that the rate of sustained virologic response in each of the four treatment groups would be higher than an adjusted historical rate of 25%, which was based on the expected response rate for this patient population (see the Supplementary Appendix). We calculated that a sample of 100 patients in each treatment group would provide the study with more than 99% power to detect an improvement of at least 45 percentage points in the rate of response over the historical null rate of 25%, with the use of a two-sided, exact one-sample binomial test and a significance level of 0.0125 on the basis of a Bonferroni correction. An exact logistic-regression analysis was performed to identify baseline factors associated with sustained virologic response; the Kruskal-Wallis test was used to test for the overall difference across treatment groups for continuous variables, and the Cochran-Mantel-Haenszel test was used for categorical variables. See the Supplementary Appendix for methods used in the regression analyses.
Results
Baseline Characteristics
Of the 551 patients who underwent initial screening, 441 underwent randomization and 440 began treatment (Table S1 and Figure S2 in the Supplementary Appendix). The demographic and baseline clinical characteristics of the patients were generally balanced among the four treatment groups (Table 1). As expected in a population of patients who had not had a sustained virologic response to interferon-based therapy, most patients (88%) had the non-CC IL28B genotype. A total of 20% of the patients in each of the four treatment groups had cirrhosis. Overall, 52% of the enrolled patients had received prior treatment with a protease-inhibitor regimen.
Efficacy
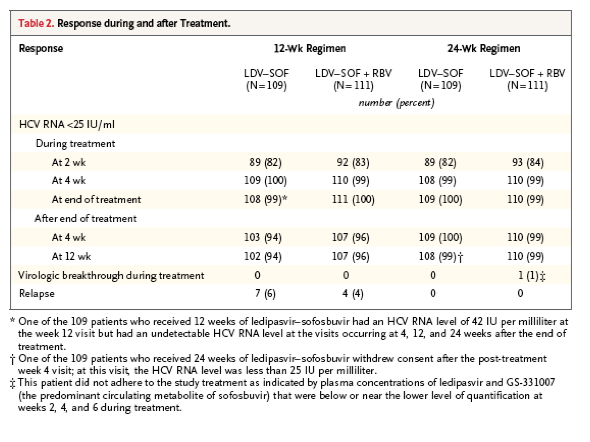
In all four treatment groups, the rate of sustained virologic response was superior to the adjusted historical response rate of 25% (P<0.001 for all comparisons). The rates of sustained virologic response at 12 weeks after the end of treatment were as follows: among 109 patients who received 12 weeks of ledipasvir-sofosbuvir, 102 had a sustained virologic response (94%; 95% confidence interval [CI], 87 to 97); among 111 who received 12 weeks of ledipasvir-sofosbuvir plus ribavirin, 107 had a sustained virologic response (96%; 95% CI, 91 to 99); among 109 who received 24 weeks of ledipasvir-sofosbuvir, 108 had a sustained virologic response (99%; 95% CI, 95 to 100); and among 111 who received 24 weeks of ledipasvir-sofosbuvir plus ribavirin, 110 had a sustained virologic response (99%; 95% CI, 95 to 100) (Table 2). Across treatment groups, 85 to 91% of the patients with an elevated serum level of alanine aminotransferase at baseline had a reduction to a level that was within the limits of the normal range by week 2 of treatment (Table S4 in the Supplementary Appendix).
Overall, 11 of the 440 patients (2%) had a virologic relapse after the end of treatment: 7 of 109 patients (6%) in the group that received 12 weeks of ledipasvir-sofosbuvir and 4 of 111 (4%) in the group that received 12 weeks of ledipasvir-sofosbuvir plus ribavirin. A total of 10 of these 11 patients had a relapse by week 4 after the end of treatment; 1 patient had a relapse between post-treatment weeks 4 and 12. The characteristics of the patients who had a relapse are shown in Table S5 in the Supplementary Appendix.
No patient in a group that received 24 weeks of treatment had a virologic relapse. Only two patients who received 24 weeks of treatment did not have a sustained virologic response: one patient who received ledipasvir-sofosbuvir withdrew consent after post-treatment week 4 (HCV RNA level at post-treatment week 4, <25 IU per milliliter), and one who received ledipasvir-sofosbuvir plus ribavirin had virologic rebound during treatment. This patient had low-to-undetectable plasma concentrations of ledipasvir and GS-331007 (the major circulating metabolite of sofosbuvir) at weeks 2, 4, and 6, which suggested nonadherence to the study-drug regimen.
All 427 patients who had a sustained virologic response at 12 weeks after the end of treatment also had a sustained virologic response at 24 weeks after the end of treatment. No patient had a relapse after post-treatment week 12.
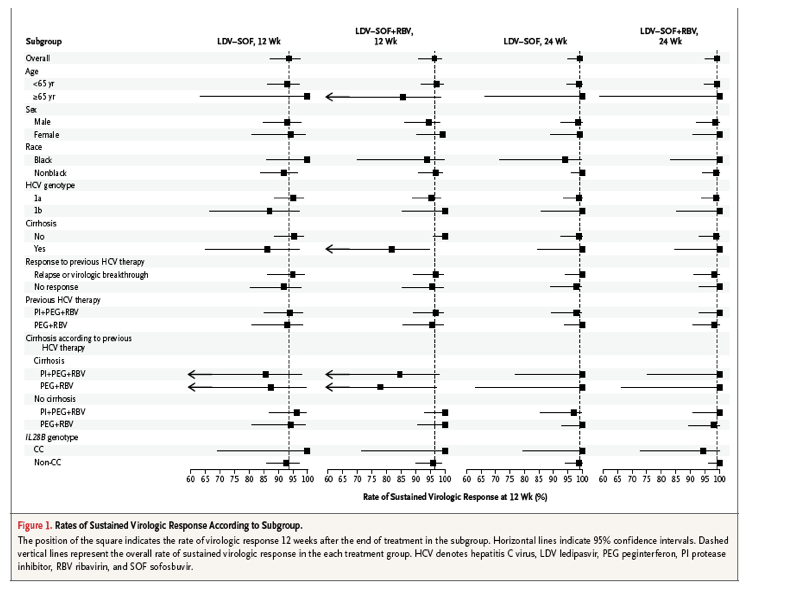
The rates of sustained virologic response in various patient subgroups are shown in Figure 1 and in Table S3 in the Supplementary Appendix. In all the groups, the response rates were similar among patients with HCV genotype 1a infection and those with HCV genotype 1b infection, among patients who had previously received peginterferon and ribavirin and those who had received a protease-inhibitor regimen, and among patients with no response to prior treatment and those with prior virologic breakthrough or relapse. Ribavirin had no effect on response rates, regardless of treatment duration.
Among patients with cirrhosis who were assigned to 12 weeks of treatment, the rates of sustained virologic response were 86% for those who received ledipasvir-sofosbuvir and 82% for those who received ledipasvir-sofosbuvir plus ribavirin; the respective rates among patients without cirrhosis were 95% and 100%. Among patients assigned to receive 24 weeks of treatment, the response rates were similar for patients with cirrhosis and those without cirrhosis. The difference between the rates of response among patients with cirrhosis who received 12 weeks of treatment and the rates among patients with cirrhosis who received 24 weeks of treatment was significant (P=0.007 by the stratified Cochran-Mantel-Haenszel test).
The multivariate exact logistic-regression analysis identified the absence of cirrhosis as the only baseline factor associated with a significant increase in the rate of response (Tables S6 and S7 in the Supplementary Appendix). Overall, the rate of sustained virologic response was 98% (95% CI, 96 to 99) among patients without cirrhosis and 92% (95% CI, 84 to 97) among those with cirrhosis. We explored whether differences in the kinetics of virologic suppression during early treatment among patients with cirrhosis and those without cirrhosis could predict the differences in response rates. Viral kinetics during the first 2 weeks of treatment did not predict the treatment outcome with ledipasvir-sofosbuvir, regardless of cirrhosis status (Tables S9 and S10 in the Supplementary Appendix). No baseline factors predictive of relapse in patients with cirrhosis were identified.
Virologic Resistance Testing
At baseline, variants associated with resistance to NS5A inhibitors were detected by means of deep sequencing in 62 of the 439 patients (14%) for whom data were available; 55 of the 62 patients (89%) had a sustained virologic response. Variants associated with resistance to NS3/4A protease inhibitors were detected at baseline in 163 of the 228 patients (71%) who underwent successful sequencing and had received prior treatment with a protease-inhibitor regimen; 159 of the 163 patients (98%) had a sustained virologic response.
Of the 7 patients who received 12 weeks of ledipasvir-sofosbuvir and had a relapse, 4 (57%) had NS5A-resistant variants at baseline. Of the 4 patients who received 12 weeks of ledipasvir-sofosbuvir plus ribavirin and had a relapse, 2 (50%) had NS5A-resistant variants at baseline. All 11 patients who had a relapse had detectable NS5A-resistant variants at the time of the relapse. Patients with NS5A-resistant variants at baseline and those without NS5A-resistant variants at baseline had similar viral kinetics during early treatment (Figure S3 and S4 in the Supplementary Appendix). The NS5B-resistant variant S282T was not detected in any patient at baseline or at any time thereafter.
Safety
The majority of patients in each treatment group (67 to 90%) had adverse events, most of which were mild to moderate in severity. None of the 440 patients in the study discontinued treatment prematurely owing to adverse events. No patient in either 12-week group had a serious adverse event. Among the patients assigned to a 24-week regimen, the rate of serious adverse events was 6% among those who received ledipasvir-sofosbuvir and 3% among those who received ledipasvir-sofosbuvir plus ribavirin (P=0.36). Table 3.
Among patients who received ledipasvir-sofosbuvir alone, the incidence of adverse events was higher in the 24-week group than in the 12-week group (81% vs. 67%). Among those who received ledipasvir-sofosbuvir plus ribavirin, the incidence of adverse events was similar in the 12-week group and the 24-week group (86% and 90%, respectively). Patients in the groups that received ribavirin had higher rates of events that are known to be associated with ribavirin therapy14 - fatigue, nausea, insomnia, arthralgia, cough, rash, irritability, dyspnea, and anemia - than did the patients who received ledipasvir-sofosbuvir alone (Table 3).
The mean change in the hemoglobin level from baseline to week 12 was -0.5 g per deciliter and -0.6 g per deciliter in patients who received 12 weeks and 24 weeks, respectively, of ledipasvir-sofosbuvir, as compared with -2.5 g per deciliter and -2.4 g per deciliter in patients who received 12 weeks and 24 weeks, respectively, of ledipasvir-sofosbuvir plus ribavirin. Mild-to-moderate (grade 1 or 2) hyperbilirubinemia developed in more patients who received ledipasvir-sofosbuvir plus ribavirin (in 32% of patients who received 12 weeks of therapy and 41% of those who received 24 weeks of therapy) than in those who received only ledipasvir-sofosbuvir (1% and 7%, respectively). Two patients who received ledipasvir-sofosbuvir plus ribavirin had grade 3 hyperbilirubinemia. The rates of laboratory abnormalities were otherwise similar among the four treatment groups. Platelet counts and albumin levels during treatment are shown in Tables S11 and S12 in the Supplementary Appendix.
Discussion
In this study, treatment with the once-daily, single-tablet regimen of ledipasvir-sofosbuvir resulted in a sustained virologic response in 94 to 99% of patients with HCV genotype 1 infection who had not had a sustained virologic response after prior interferon-based treatment, including protease-inhibitor regimens. These rates of response are among the highest reported to date for HCV genotype 1 infection. The rates of sustained virologic response were similar, with widely overlapping confidence intervals, among patients who received 12 weeks of treatment and among those who received 24 weeks of treatment, and also among those who received ledipasvir-sofosbuvir alone and among those who received ledipasvir-sofosbuvir plus ribavirin. However, this study was not powered to compare responses to regimens with and without ribavirin or to 12 weeks and 24 weeks of treatment.
Ledipasvir-sofosbuvir was not associated with any new or characteristic adverse events, although the patients who received ledipasvir-sofosbuvir plus ribavirin had higher rates of fatigue, nausea, insomnia, arthralgia, cough, rash, irritability, dyspnea, and anemia - events that are consistent with the known side effects of ribavirin - than did those who received ledipasvir-sofosbuvir alone.14 Overall, the rate of adverse events was substantially lower in the group that received 12 weeks of ledipasvir-sofosbuvir alone (67%) than in the other three treatment groups (81 to 90%).
The study population included substantial numbers of patients with characteristics historically associated with a poor response to treatment.15 In particular, 84 to 91% of the patients had a non-CC IL28B genotype, 41 to 46% had a documented prior nonresponse to interferon-based therapy, and 52% had previously been treated with a protease-inhibitor regimen and were therefore without treatment options. This treatment was effective in patients who had not had a sustained virologic response with a protease-inhibitor regimen - a population that accounts for a substantial proportion of patients with diagnosed HCV infection. The rates of sustained virologic response in all these difficult-to-treat subgroups ranged from 92% to 100%.
In the 12-week treatment groups, patients with cirrhosis had modestly lower rates of response (86% with ledipasvir-sofosbuvir and 82% with ledipasvir-sofosbuvir plus ribavirin) than did those without cirrhosis (95 and 100%, respectively), whereas in the 24-week treatment groups, the rates of response were similar in patients with cirrhosis (99% with both regimens) and those without cirrhosis (100% with both regimens). However, this observation is preliminary, since the study was not powered for intergroup comparisons. We explored the usefulness of virologic suppression during early treatment in predicting sustained virologic response in patients with cirrhosis. Among patients with cirrhosis who received 12 weeks of ledipasvir-sofosbuvir alone, 80% of those who had a detectable HCV RNA level at week 2 had a sustained virologic response (Table S10 in the Supplementary Appendix), indicating that virologic response during early treatment does not accurately identify the small subset of patients who might benefit from 24 weeks of treatment. We were unable to identify any baseline characteristics that might predict which patients with cirrhosis were most likely to have a relapse after 12 weeks of treatment.
The NS5B S282T variant, which reduces susceptibility to sofosbuvir, was not observed in this study, confirming the high genetic barrier to the development of resistance that has been observed in previous studies of sofosbuvir. Treatment failure with inhibitors of the HCV NS5A protein is often associated with the presence at baseline of NS5A variants that are resistant to treatment and also with the rapid development of resistant variants during treatment. Although no virologic relapse was observed in patients who received 24 weeks of treatment, 11 of those who received 12 weeks of treatment had a relapse. Of the 11 patients who had a relapse after treatment, 6 had NS5A-resistant variants at baseline. All the patients who had a relapse had detectable NS5A-resistant variants at the time of virologic failure. Early reductions in the HCV RNA level were similar in patients without resistant variants at baseline and in those with resistant variants at baseline, including those who had a relapse.
The study also characterizes the burden of adverse events associated with ribavirin. Higher rates of constitutional and neuropsychiatric side effects were observed in the two groups that received the ribavirin-containing regimen than in the two groups that received ledipasvir-sofosbuvir alone. Decreases in the hemoglobin level and increases in the bilirubin level were seen in the two groups that received the ribavirin-containing regimen - findings consistent with ribavirin-mediated hemolysis - but not in the two groups that received ledipasvir-sofosbuvir alone. The exclusion of patients who had discontinued prior therapy owing to adverse events, which was intended to restrict the study population to patients who had not had a response to prior therapy, may have inadvertently selected for patients who were more likely to have a low rate of adverse events.
In conclusion, treatment with a single-tablet regimen containing ledipasvir and sofosbuvir resulted in high rates of sustained virologic response among patients with HCV genotype 1 infection who had not had a response to prior interferon-based treatment.
|
| |
|
|
|
|
|