| |
CUPIC & Adverse Events Study & Commentary
|
| |
| |
Download The PDF here
Download The PDF here
Download The PDF here
Reply to - From the CUPIC study: Great times are not coming (?)
Journal of Hepatology
Dec 30 2013
Christophe Hezode, Helene Fontaine, Yoann Barthe, Fabrice Carrat, Jean-Pierre Bronowicki
"In summary, we have clearly identified a subgroup of patients with a high risk of severe complications raising the question as to whether these patients, who are the most in need of therapy, should be treated with triple therapy including boceprevir or telaprevir. In practice, the risk of developing severe complications or death should be carefully balanced against the likelihood of a virological response and subsequent improvement of survival. These patients could benefit from prophylaxis (antibiotics) of treatment complications and should undergo careful monitoring while on therapy. Alternatively, most of them can wait for all -oral, IFN-free regimens."
To the Editor
Schmidt-Martin D and colleagues raised one comment (see comment below) regarding the main recommendation of our article meaning that patients with hypoalbuminemia (<35 g/dl) and thrombocytopaenia (˛100,000/mm3) should not be treated with an interferon based triple therapy due to the high risk to develop severe complications during the first 16 weeks of therapy [1]. As mentioned inthe supplementary materials,severe complications were defined as any of death, grade 3 or 4 infection, hepatic decompensation. The cause of death, the description of severe infections and hepatic decompensation were detailed in the results section. A severe infection (grade 3 or 4) was defined by an infectious episode needing hospitalisation or life-threatening. Among the 6 patients who died during the first weeks of treatment, 3 had hypoalbuminemia (<35 g/dl) and thrombocytopenia (˛100,000/mm3) and the 3 others either hypoalbuminemi a or thrombocytopenia.
CUPIC was a cohort in setting of the French early access program explaining that all patients received antiviral therapy and the lack of control group. In five recent studies (including a meta-analysis) evaluating the natural history of cirrhotic patients with characteristics comparable to those from the CUPIC cohort, the annual incidence of deaths and/or liver decompensation varied from 6.2 to 11% on treatment, challenging the indication for triple therapy in this subgroup of patients [2-6]. In the subgroup of patients who combined both predictors of severe complications (albumin <35 g/dl and platelet count ˛100,000/mm3), their occurrence was 44.1% during the first 16 weeks. We cannot exclude that this rate could increase overtime in patients receiving 48 weeks of therapy. This result strongly suggests that the risk to develop severe complications is higher in patients with both predictors treated with triple therapy compared to untreated cirrhotic patients. We agree that the lack of a control group does not allow us to distinguish between severe events caused by treatment from those caused by the cirrhosis itself. However, based on comparisons with other studies in cirrhotic patients, the rate of observed severe events was much higher in this particular group of patient with advanced cirrhosis, low platelet count and low albumin level. Moreover severe infection is a relatively rare event in non-treated compensated viral cirrhosis.
Therefore, it's likely that the treatment contributed to these events. We agree that international treatment guidelines [7-8] recommend starting triple therapy in HCV genotype 1 patients with compensated cirrhosis. However, these guidelines were based on efficacy and safety data reported in cirrhotic patients included in phase III clinical trials. A crucial point is that low platelets count and hypoalbuminemia were exclusion criteria for these studies, meaning that no safety data was available in such patients at the time of international guidelines. Moreover, it is important to note that patients included in the international telaprevir early access program patients had severe fibrosis (n=741, Metavir F3 or Ishak 3-4) or compensated cirrhosis (n=840, Child-Pugh Grade A) and no history of decompensated liver disease. Additionally, patients must have a platelet count of at least 90,000/mm3 and albumin >35 g/L [9). This study does not allow providing safety data in patio nts with both predictors of severe complications. Schmidt-Martin D and colleagues suggest that the poor safety profile in such patients could be explained by the large number of centres involved in the CUPIC cohort with variation in the treatment and monitoring protocols. However, there was no impact of the experience of the centre, evaluated by the number of treated patients (<5 vs. ł 5 patients) on the occurrence of severe complications and grade 3/4 anaemia (Hb <8 g/dl) or blood transfusion.
In summary, we have clearly identified a subgroup of patients with a high risk of severe complications raising the question as to whether these patients, who are the most in need of therapy, should be treated with triple therapy including boceprevir or telaprevir. In practice, the risk of developing severe complications or death should be carefully balanced against the likelihood of a virological response and subsequent improvement of survival. These patients could benefit from prophylaxis (antibiotics) of treatment complications and should undergo careful monitoring while on therapy. Alternatively, most of them can wait for all -oral, IFN-free regimens.
--------------------------------------------------
Jnl of Hepatology
Article in Press
Accepted Manuscript
unedited manuscript that has been accepted for publication
Received 18 November 2013; accepted 22 November 2013. published online 26 December 2013.
Letter to the Editor
From the CUPIC study: Great times are not coming (?)
Daniel Schmidt, Aidan McCormick, Diarmaid Houlihan
We note, with interest, the findings of the interim results from the CUPIC study reported recently in the Journal of Hepatology[1]. The authors demonstrate a high incidence of serious adverse events (40.0%), and of death and severe complications (severe infection or hepatic decompensation) (6.4%), in a difficult to treat cohort of patients. Most notably however, the authors have proposed that compensated cirrhotic patients with chronic HCV with both thrombycytopaenia (Platelets <100,000) hypoalbuminaemia (<35 g/dL) should not be treated with triple therapy.
There are a number of important considerations for accurate interpretation of the data. Although acknowledged by the authors, it is important to re-emphasise that CUPIC lacked a control group. The author's conclusions must, therefore, be balanced against the published natural history of HCV cirrhotic patients. The benefits of sustainedvirology response (SVR) in this group are well documented [2]. Hence, international treatment guidelines generally recommend antiviral therapy in this patient group [3, 4]. Indeed, in controlled trials of dual therapy of patients with decompensated cirrhosis, the treatment group had more favourable outcomes compared to control group [5]. Additionally, treatment of HCV patients on the liver transplant waiting list does not appear to increase overall mortality compared with untreated patients [6]. Thus, the benefits of achieving SVR, in this difficult population, cannot be overstated [7].
CUPIC was not designed or powered for interim safety analysis. Indeed recent data investigating the therapeutic potential of triple therapy in patients with severe fibrosis or cirrhosis demonstrated lower complication rates than those observed in CUPIC[8, 9].
Possible explanations for this difference include multiple treatment centres (n=56) with variation in the treatment and monitoring protocols. In addition 'severe infection' is not clearly defined in the manuscript and this was the most commonly (24 of 32 cases) reported severe complication. Indeed, 7 of the 24 patients who survived a severe infection continued on treatment without requiring a dose reduction and a further unreported number continued treatment at a reduced dose. Deaths in the total cohort were relatively uncommon (n=6, <1%) however neither the albumin level nor platelet count is reported for these. Two of the six deaths reported occurred prior to the introduction of the protease inhibitor which, may indicate that these subjects were poor candidates for treatment. The association between platelet count and albumin level and mortality during HCV treatment is not novel. Previous work has demonstrated a per annum mortality for HCV patients with thrombocytopaenia alone (defined as platelet count <150,000 rather than the level of 100,000 used in the paper) of 4%, which would correspond to 4 deaths over the course of a year among the 103 thrombocytopaenic patients [10]. Furthermore, hypoalbuminaemia has also been independently associated with increased mortality in cirrhotic patients prompting calls for the adoption of a modified MELD score incorporating albumin[11]. It seems incontrovertible that cirrhotic patients are at greater risk of complication from triple therapy, but suggesting that platelet count (<100,000) and albumin level <35) should preclude consideration for treatment without describing the natural history of untreated HCV in a similar cohort appears unbalanced. For many of these patients, the promise shown in clinical trials of the next generation of directly acting antivirals, the safety of which in these cohorts remains also untested may well come too late.
---------------------------------------
SVR12 rates and safety of triple therapy including telaprevir or boceprevir in 485 cirrhotic non responders treated in the French Early Access Program (ANRS CO20-CUPIC)
http://www.natap.org/2013/EASL/EASL_16.htm
---------------------------------------
Triple therapy in treatment-experienced patients with HCV-cirrhosis in a multicentre cohort of the French Early Access Programme (ANRS CO20-CUPIC) - NCT01514890
Journal of Hepatology September 2013
Background & Aims
In phase III trials, the safety profile of triple therapy (pegylated interferon/ribavirin with boceprevir or telaprevir) seems to be similar in HCV treatment-experienced cirrhotic and non-cirrhotic patients, but few cirrhotics were included. We report the week 16 safety and efficacy analysis in a cohort of compensated cirrhotics treated in the French Early Access Programme.
Methods
674 genotype 1 patients, prospectively included, received 48weeks of triple therapy. The analysis is restricted to 497 patients reaching week 16.
Results
A high incidence of serious adverse events (40.0%), and of death and severe complications (severe infection or hepatic decompensation) (6.4%), and a difficult management of anaemia (erythropoietin and transfusion use in 50.7% and 12.1%) were observed. Independent predictors of anaemia <8 g/dl or blood transfusion were: female gender (OR 2.19, 95% CI 1.11-4.33, p = 0.024), no lead-in phase (OR 2.25, 95% CI 1.15-4.39, p = 0.018), age 65 years (OR 3.04, 95% CI 1.54-6.02, p = 0.0014), haemoglobin level (≤12 g/dl for females, ≤13 g/dl for males) (OR 5.30, 95% CI 2.49-11.5, p = 0.0001). Death or severe complications were related to platelets count ≤100,000/mm3 (OR 3.11, 95% CI 1.30-7.41, p = 0.0105) and albumin <35 g/dl (OR 6.33, 95% CI 2.66-15.07, p = 0.0001), with a risk of 44.1% in patients with both. However, the on-treatment virological response was high.
Conclusions
The safety profile was poor and patients with platelet count ≤100,000/mm3 and serum albumin <35g/L should not be treated with the triple therapy.
Introduction
Since 2011, the standard of care for genotype 1 chronic hepatitis C is a triple therapy combining pegylated interferon (PegIFN), ribavirin (RBV), and telaprevir (TVR) or boceprevir (BOC), NS3/4A protease inhibitors (PIs) [1], [2], [3], [4]. In the phase III studies, the sustained virological response (SVR) rate varied according to the patient status (treatment-na•ve or treatment-experienced) and severity of fibrosis. The safety profile of triple therapy with PIs in part reflects the known profile of PegIFN and RBV. However, the addition of PIs is responsible for incremental anaemia and dysgeusia with BOC, and incremental anaemia, frequent skin reactions and gastrointestinal disorders with TVR [1], [2], [3], [4]. The proportion of treatment discontinuations owing to adverse events ranged from 8% to 16% with BOC-based triple therapy and from 10% to 15% with TVR-based triple therapy in the phase III trials in both treatment-na•ve and -experienced patients [1], [2], [3], [4]. It is well established that cirrhotics are priority patients for treatment, given the high risk of hepatic decompensation, hepatocellular carcinoma, and liver-related death in this population [5], [6], [7], [8]. In these patients, viral eradication is associated with a decreased risk of progression of liver disease [9], [10], [11], [12]. The safety profile of PegIFN/RBV in compensated cirrhotic patients is compatible with its use in real-life practice [13], [14]. However, antiviral therapy increases the risk of bacterial infections in cirrhotic patients with poor liver function, awaiting liver transplantation [15]. In phase III trials, the safety profile of triple therapy was available for 139 patients with compensated cirrhosis only, and seemed to be comparable with non-cirrhotic patients [16]. However, cirrhotic patients included in these trials were highly selected and did not reflect well the actual population of cirrhotic patients infected with HCV awaiting antiviral therapy with the new standard of care.
The French "Autorisation Temporaire d'Utilisation" (ATU) is an early access programme that gives patients access to a medicinal product before marketing authorization. Patients with compensated cirrhosis who were relapsers or partial responders after prior PegIFN/RBV treatment were treated prior to approval of TVR and BOC within the framework of this programme, beginning in January 2011.
The ANRS CO20 CUPIC (Compassionate Use of Protease Inhibitors in viral C Cirrhosis) study is a cohort study sponsored by the French National Agency for Research on AIDS and Viral Hepatitis (ANRS) including patients benefiting from the ATU early access programme in selected centres. The aim of this study was to evaluate the efficacy and safety of triple combination therapy including a PI in treatment-experienced cirrhotic patients in the real-life setting of the French early access programme. We report here the week 16 interim safety and efficacy results.
Patients and methods
Patients
The ANRS CO20-CUPIC cohort (ClinicalTrials.gov number NCT01514890) is a national multicentre prospective cohort study conducted in 56 French centres. From February 2011 to April 2012, patients with compensated cirrhosis (Child-Pugh class A) chronically infected with HCV genotype 1, who did not achieve an SVR after a prior course of IFN-based therapy and who started triple therapy, were recruited. Initially, only relapsers and partial responders were eligible in the French early access programme. Since the approval of both PIs, the inclusion criteria were amended in September 2011 allowing the inclusion of null responders (Supplementary Material).
The diagnosis of cirrhosis was made by liver biopsy or non-invasive tests, FibrotestTM or Fibroscan¨ or Fibrometer¨ or Hepascore¨, at the discretion of the investigator, according to the French recommendations [17]. Patients with HIV or HBV co-infection, renal insufficiency (defined by creatinine clearance <50ml/min) or organ graft were not eligible for inclusion.
Written informed consent was obtained from each patient before enrolment. The protocol was conducted in accordance with the Declaration of Helsinki and French law for biomedical research and was approved by the "Ile de France IX" Ethics Committee (CrŽteil, France).
Objectives
The main goal of this interim analysis was to evaluate safety and tolerability among patients who received at least 16 weeks of antiviral treatment, i.e., at least 12 weeks of PI in combination with PegIFN and RBV. The secondary objective was to assess the on-treatment virologic response at week 16.
Treatments
Treatment was prescribed at the discretion of each investigator without randomization, which precludes any comparison between the two treatment regimens. The treatment schedules and recommended stopping rules are detailed in the Supplementary Material.
Safety assessments
The safety profile assessment is described in Supplementary Material. Data on all adverse events were collected until the 16th week. Clinical and laboratory grade 3 or 4 adverse events, serious adverse event (SAE), and serious cutaneous adverse reaction (SCAR) [18] are defined in Supplementary Material. Moreover, grade 2 anaemia was also recorded. Anaemia was managed at the discretion of the investigator, consisting of RBV dose reductions and/or erythropoietin (EPO) administration, authorized in France when Hb is below 10g/dl, and/or blood transfusion. The use of other hematopoietic growth factors related to neutropenia and thrombocytopenia was also collected.
HCV-RNA level monitoring
HCV-RNA levels were measured at baseline and at weeks 4, 8, 12, 16, 24, 36, and 48 of therapy, and 12 and 24 weeks after its withdrawal, with a real-time PCR-based assay, either COBAS AmpliPrep¨/COBAS TaqMan¨ (Roche Molecular Systems, Pleasanton, California) with a lower limit of detection of 15 IU/ml, or m2000SP/m2000RT (Abbott Molecular, Des Moines, Illinois), with a lower limit of detection of 12 IU/ml. Both assays have been validated for their accuracy in patients infected with HCV genotype 1 [19], [20]. In this interim analysis, monthly on-treatment virological responses until week 16 are presented.
Statistical analysis
We estimated that 900 patients would be needed for the cohort to have a 3% precision in assessing the SVR. The safety and efficacy interim analysis was not prespecified in the protocol, but decided by the scientific committee in February 2012, based on preliminary reports of safety findings. Therefore, no sample size was planned. All patients who reached 16weeks of treatment by May 1st, 2012 were included in the analysis.
We used logistic regression models to identify predictors of severe complications and grade 3/4 anaemia or blood transfusion.
Blood parameters were categorized according to thresholds used to define eligibility in a randomized trial of protease inhibitor-based triple therapy [2]. For each tested covariate, a univariate model was estimated. Covariates with p <á05 in likelihood ratio testing in univariate analysis were included in a multivariate model, and selection of independent covariates was based on a backward elimination procedure, retaining covariates with p <á05.
Efficacy analyses were performed on an intent-to-treat basis. Missing virological measurements were imputed as treatment failures.
Comparisons between independent groups used the Mann-Whitney test or Fisher's exact test and within-group comparisons were made using the Wilcoxon signed-rank test or McNemar's Chi square tests. Proportions of adverse events across MELD groups were compared using the Cochran-Armitage Chi square for trend. All statistical computations were performed using SAS software version 9.3 (SAS Institute Inc., Cary, North Carolina).
Results
Patient characteristics
Six hundred and seventy-four patients 18 years of age were included in 56 centres, 497 of whom were included in this interim analysis, 292 treated with TVR and 205 with BOC (Fig. 1). Baseline characteristics of patients are shown in Table 1. Of these 497 patients, 337 (67.8%) were male, with a mean age of 57.1 (±9.7) years. Prior treatment response was relapse, partial response, null response, and undetermined in 223 (44.9%), 226 (45.5%), 33 (6.6%) and 15 (3.0%) patients, respectively. At the beginning of treatment, cirrhosis was compensated and classified as Child-Pugh A in all patients, except in 8 (1.6%) and 7 (1.4%) patients in whom the classification was Child-Pugh B or undetermined, respectively. Three (0.6%) patients had a history of ascites. The baseline MELD score was available in 427 (85.9%) patients and the mean value was 8.1 (±2.9). The MELD score was <10 in 350 (82.0%) patients, from 10 to 13 in 52 (12.2%) and 13 in 25 (5.8%). Upper gastrointestinal endoscopy was performed before the start of the treatment in 249 patients (50.1%) and oesophageal varices were observed in 89 (35.7%) of them. The baseline haematological characteristics were as follows: mean neutrophil count 3.3 x 103/mm3 ± 1.3; mean platelet count 150,000/mm3 ± 67,100 and mean Hb 14.7 g/dl ± 1.6. Based on age and laboratory parameters, 155 (31.2%) patients had at least one exclusion criterion for the REALIZE study [4] and 215 (43.3%) for the RESPOND-2 study [2]. Thirty-seven (7.4%) patients were diabetic. The mean baseline HCV-RNA level was 6.0 (±0.9) log10 IU/ml, and the HCV-RNA level was higher than 800,000 IU/ml in 312 (62.8%) patients with available data. The distribution of HCV genotype 1 subtype was 1a, 1b, and others (undetermined and missing data) in 179 (36.0%), 262 (52.7%) and 56 (11.3%) patients, respectively. The mean daily RBV dose was 14.3 ± 2.3 mg/kg in the global population: 14.1 ± 2.4 for patients receiving BOC and 14.4 ± 2.3 for patients receiving TVR.
Adverse events
Table 2 illustrates the safety profile of the triple therapy with TVR or BOC.
Incidence of SAEs
A high incidence of SAEs (n=493) was observed. SAEs occurred in 199 (40.0%) patients, leading to early treatment discontinuation in 58 (11.7%).
Deaths and severe complications
Deaths occurred in 6 patients during the course of therapy, mainly related to severe infections: septicaemia (n = 2), pneumonia (n = 2) and endocarditis (n = 1). The remaining death was due to hepatic decompensation related to variceal bleeding.
Death occurred after a median time of 6.4 weeks and two cases occurred during the lead-in phase, i.e., without PI. Apart from death, severe complications, including severe infections and hepatic decompensation, occurred in 32 (6.4%) patients. Severe infections were reported in 24 (4.8%) patients: pulmonary infection (n = 8), septicaemia of unidentified origin (n = 7), acute pyelonephritis (n = 4), endocarditis (n = 2), food poisoning (n = 1), cutaneous infection (n = 2). Bacteria were identified in 17 patients: Staphylococcus (n = 8), Escherichia coli (n = 5), Klebsiella (n = 1), Pyocyanic (n = 1), Bacteroides fragilis (n = 1) and Pneumococcus (n = 1). These infectious complications occurred after a median duration of antiviral treatment of 8.6 weeks (2.3-15.9). PegIFN dose was reduced or discontinued in 13 patients (54.2%) before the occurrence of severe infection, and in an additional 6 patients (25%) after severe infection onset.
In addition, episodes of hepatic decompensation were observed in 12 (2.4%) patients: ascites in 7 (1.4%), encephalopathy in 3 (0.6%), and variceal bleeding in 1 (0.2%). Overall, 32 (6.4%) patients experienced severe complications that occurred after a mean duration of 8.9 weeks (0.3-15.9), including 2 (0.4%) during the lead-in phase. Interestingly, the mean MELD score significantly increased between baseline and week 12 on both treatment regimens (with TVR and BOC, respectively), from 8.1 ± 2.8 to 8.6 ± 2.7 (p <0.0001), in 335 patients with available MELD score.
Anaemia
The incidence of anaemia (Hb ≤9.0 g/dl) was 29.4% (146 of 497), 252 (50.7%) patients received EPO, the RBV dose was reduced or discontinued in 80 (16.1%) patients, and blood transfusions were needed in 60 (12.1%) (Table 2).
Other
Grade 3 or 4 neutropenia and thrombocytopenia and early discontinuations related to these adverse events were rarely reported (Table 2). Skin disorders were not more frequent than in clinical trials and no SCAR was reported during the study period.
Factors associated with side effects
In univariate analysis, factors associated with severe complications and grade 3/4 anaemia (Hb <8g/dl) or blood transfusion are shown in Table 3.
Table 3. Univariate and multivariate analysis.
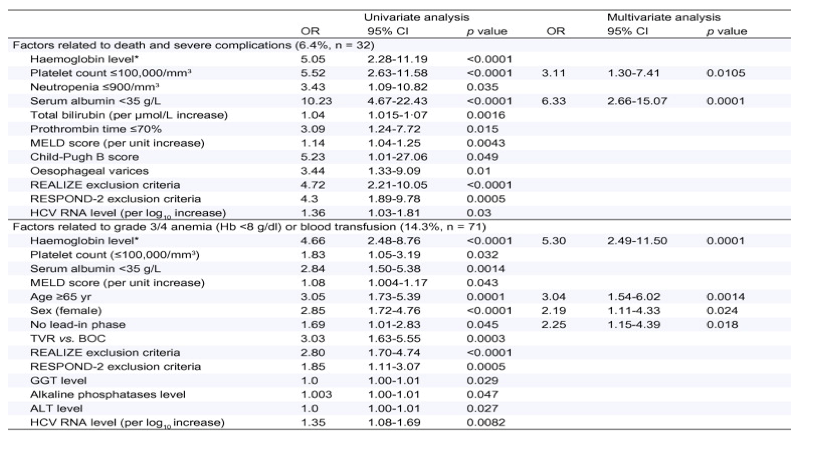
*Haemoglobin level: ≤12g/dl for females and ≤13g/dl for males.
Multivariate stepwise logistic-regression analysis served to identify four independent baseline factors as independent predictors of grade 3/4 anaemia (Hb <8 g/dl) or blood transfusion that occurred in 71 patients (14.3%): female gender (OR = 2.19, 95% CI 1.11-4.33, p = 0.024), no lead-in phase (OR = 2.25, 95% CI 1.15-4.39, p = 0.018), age 65 years (OR = 3.04, 95% CI 1.54-6.02, p = 0.0014), baseline Hb ≤12 g/dl for females and ≤13 g/dl for males (OR = 5.30, 95% CI 2.49-11.50, p = 0.0001).
In multivariate analysis, two baseline predictors of death and severe complications that occurred in 32 patients (6.4%) were: platelet count ≤100,000/mm3 (OR = 3.11, 95% CI 1.30-7.41, p = 0.0105) and serum albumin <35 g/dl (OR = 6.33, 95% CI 2.66-15.07, p = 0.0001). Using the combination of the values of these 2 predictors, the risk varied from 3.4 to 44.1% (Table 4).
Table 4. Risk of occurrence of death or severe complications according to serum albumin level and platelet count during the first 16 weeks of therapy.*

*
Baseline albumin and platelet count were available in 429 patients (missing data in 61 patients for albumin and 21 patients for platelet count). Twenty-nine cases of death or severe complications were reported and analysed in these 429 patients.
There was no impact of the experience of the centre, evaluated by the number of treated patients (<5 vs. 5 patients) on the occurrence of severe complications and grade 3/4 anaemia (Hb <8g/dl) or blood transfusion.
Treatment virological response
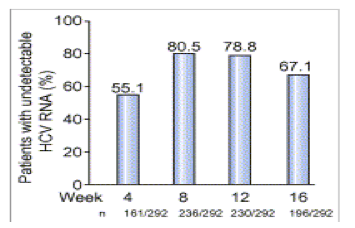
In intent-to-treat analysis, among the 292 patients treated with TVR, HCV-RNA was undetectable in 161 (55.1%), 236 (80.5%), 230 (78.8%), and 196 (67.1%) at weeks 4, 8, 12, 16, respectively. Of these, in the 42 (14.4%) patients treated with a lead-in phase, these percentages were 4 (9.5%), 32 (76.2%), 30 (71.4%), and 25 (59.5%), respectively (Fig. 2). In the 250 patients treated without lead-in phase, these percentages were 157 (62.8%), 204 (81.6%), 200 (80.0%), 171 (68.4%), respectively. At week 16, response rate was significantly higher in relapsers (74.8%) than in partial responders (66.2%) and in null responders (45.8%), p=0.005.
In patients treated with BOC, virological response was achieved in 2.4 (5 of 205), 37.6 (77 of 205), 54.6 (112 of 205), and 58.0% (118 of 205) of cases, at weeks 4, 8, 12, 16, respectively (Fig. 3). At week 16, response rate was significantly higher in relapsers (69.0%) than in partial responders (50.0%) and in null responders (22.2%), p=0.001.
Discussion
The CUPIC cohort is the largest cohort of treatment-experienced cirrhotic patients infected with genotype 1 and treated with BOC or TVR triple therapy in the real-life setting. Importantly, the choice of antiviral treatment was made by physicians, and patients were not randomized, which precludes any comparison between the two PIs. However, for each drug regimen, our results provide a good reflection of the safety profile in this specific population, with a clear indication for antiviral therapy that was not included in phase II and phase III trials.
The safety profile was poor for treatment regimens including both PIs, mainly because of a high number of SAEs and the occurrence of deaths and severe complications, such as severe infection or hepatic decompensation in 6.4% of the patients. These severe complications were not previously reported in treatment-experienced cirrhotic patients included in phase III clinical trials [16]. This major concern could be explained by the difference of baseline characteristics of patients between our real-life cohort and selected patients enrolled in phase III clinical trials. Our patients had at least one exclusion criterion for REALIZE and RESPOND-2 in 31.2% and 43.3% of the cases, respectively. Our patients who received TVR compared with cirrhotic patients enrolled in REALIZE [16] were older (57.2 vs. 54.0 years), with a lower mean Hb level (14.6 vs. 15.6 g/dl) and a lower mean platelet count (152,000 vs. 167,000/mm3), suggesting more advanced liver disease. Limited data were available in cirrhotic patients enrolled in the RESPOND-2 study with younger patients (54.9 vs. 57.2) [21]. However, our patients had compensated liver disease and the vast majority had no advanced cirrhosis, as suggested by a baseline Child-Pugh score A and a MELD score <13 observed in 98.4% and 93.9% of them, respectively.
In multivariate analysis, two baseline predictors of severe complications were platelet count ≤100,000/mm3 and serum albumin <35 g/L. The combination of both allowed us to define a subgroup of patients (representing 7.9% (34/429 patients) of the population with both predictors available) with a high risk (44.1%) of severe complications, suggesting that they should not be treated with triple therapy. In the others (92.1%), the risk was lower (≤7.1%), suggesting that these patients should be treated because the high on-treatment virological response rate may translate into a high SVR rate, which may in turn decrease morbidity and mortality [9], [10], [11].
It has already been shown that severe complications are associated with IFN-based treatment in patients with decompensated or Child-Pugh B-C cirrhosis [15], [22]. It is currently difficult to know if the severe complications are mainly associated with PegIFN/RBV and/or with PI use in these patients with advanced cirrhosis. Since two deaths occurred during the lead-in phase, one might hypothesize that severe complications resulted, at least in part, from IFN/RBV administration.
Despite the large use of EPO in our cohort, the incidence of severe anaemia was high and its management was unusually difficult, leading to a high frequency of blood transfusions. In REALIZE and RESPOND-2, the rate of anaemia was slightly higher in cirrhotic than in non-cirrhotic patients [4], [21]. For the overall treatment duration (48 weeks), EPO was used in 41% to 46% of BOC recipients [4], [21], whereas EPO was prescribed in 53.8% with TVR and 46.3% with BOC during the first 16 weeks of triple therapy in our cohort. Blood transfusions were needed in 16.1% of cases with TVR and 6.3% with BOC, percentages that are unexpectedly high compared with phase III clinical trials.
Independent predictive factors related to anaemia (Hb <8 g/dl) or blood transfusion were female gender, age 65 years, the absence of lead-in phase, and low baseline Hb level. The positive impact of the lead-in phase on anaemia during the first 16 weeks of treatment could be explained by a shift in the maximum Hb decrease. It will be interesting to evaluate the impact of the lead-in phase on anaemia for the overall duration of antiviral treatment.
We acknowledge that in the absence of control groups, it was impossible to correctly assess the benefit risk, as all complications observed in the treated patients are also observed in untreated severe cirrhotic patients [23], [24].
Trials with different combinations of less toxic direct antiviral agents with additive potency may provide new IFN-free regimens with increased SVR, and better safety profile in cirrhotics [25].
In conclusion, this cohort study provides new clinical information for the management of triple therapy in cirrhotic patients and shows that the safety profile of TVR- and BOC-based triple therapy is poor in a real-life setting. The rate of SAEs, including death, severe infection, hepatic decompensation, and difficult-to-treat anaemia, was high, but was associated with high rates of on-treatment virological response. Serum albumin level and platelet count should be evaluated to determine the risk/benefit ratio of triple therapy in cirrhotic patients and to decide treatment. Treatment-experienced patients with compensated cirrhosis combining a platelet count ≤100,000/mm3 and serum albumin <35 g/L should not be treated with a triple combination. IFN-free regimens need to be evaluated in this situation. The other patients could be treated cautiously and carefully monitored.
|
|
| |
| |
|
|
|