| |
All-oral daclatasvir plus asunaprevir for hepatitis C virus genotype 1b: a multinational, phase 3, multicohort study
|
| |
| |
Download the PDF here
The Lancet, Early Online Publication, 28 July 2014
Michael Manns, Stanislas Pol, Ira M Jacobson, Patrick Marcellin, Stuart C Gordon, Cheng-Yuan Peng, Ting-Tsung Chang, Gregory T Everson,
Jeong Heo, Guido Gerken, Boris Yoff e, William J Towner, Marc Bourliere, Sophie Metivier, Chi-Jen Chu, William Sievert, Jean-Pierre Bronowicki,
Dominique Thabut, Youn-Jae Lee, Jia-Horng Kao, Fiona McPhee, Justin Kopit, Patricia Mendez, Misti Linaberry, Eric Hughes, Stephanie Noviello, on
behalf of the HALLMARK-DUAL Study Team
"We did this phase 3, multicohort study (HALLMARK-DUAL) at 116 sites in 18 countries between May 11, 2012, and Oct 9, 2013. Patients were adults with chronic HCV genotype 1b infection who were treatment-naive; previous non-responders to peginterferon alfa plus ribavirin; or medically ineligible for, previously intolerant of, or ineligible for and intolerant of peginterferon alfa plus ribavirin........The treatment-naive group assigned to daclatasvir plus asunaprevir continued open-label treatment to the end of week 24; participants assigned to placebo entered another daclatasvir plus asunaprevir study. Non-responders and ineligible, intolerant, or ineligible and intolerant patients received open-label daclatasvir plus asunaprevir for 24 weeks. The primary endpoint was sustained virological response at post-treatment week 12..... Daclatasvir plus asunaprevir provided sustained virological response in 182 (90%, 95% CI 85-94) patients in the treatment-naive cohort, 168 (82%, 77-87) in the non-responder cohort, and 192 (82%, 77-87) in the ineligible, intolerant, or ineligible and intolerant cohort."
Summary
Background
An unmet need exists for interferon-free and ribavirin-free treatments for chronic hepatitis C virus (HCV) infection. In this study, we assessed all-oral therapy with daclatasvir (NS5A replication complex inhibitor) plus asunaprevir (NS3 protease inhibitor) in patients with genotype 1b infection, including those with high unmet needs or cirrhosis, or both.
Methods
We did this phase 3, multicohort study (HALLMARK-DUAL) at 116 sites in 18 countries between May 11, 2012, and Oct 9, 2013. Patients were adults with chronic HCV genotype 1b infection who were treatment-naive; previous non-responders to peginterferon alfa plus ribavirin; or medically ineligible for, previously intolerant of, or ineligible for and intolerant of peginterferon alfa plus ribavirin. Treatment-naive patients were randomly assigned (2:1 ratio) by an interactive voice-response system with a computer-generated random allocation sequence (stratified by cirrhosis status) to receive daclatasvir 60 mg once daily plus asunaprevir 100 mg twice daily or placebo for 12 weeks. Patients and investigator sites were masked to treatment assignment and HCV RNA results to the end of week 12. The treatment-naive group assigned to daclatasvir plus asunaprevir continued open-label treatment to the end of week 24; participants assigned to placebo entered another daclatasvir plus asunaprevir study. Non-responders and ineligible, intolerant, or ineligible and intolerant patients received open-label daclatasvir plus asunaprevir for 24 weeks. The primary endpoint was sustained virological response at post-treatment week 12. Efficacy analyses were restricted to patients given daclatasvir plus asunaprevir. This trial is registered with ClinicalTrials.gov, number NCT01581203.
Findings
This study included 307 treatment-naive patients (205 received daclatasvir plus asunaprevir and 102 received placebo; all randomly assigned patients received the intended treatment), 205 non-responders, and 235 ineligible, intolerant, or ineligible and intolerant patients. Daclatasvir plus asunaprevir provided sustained virological response in 182 (90%, 95% CI 85-94) patients in the treatment-naive cohort, 168 (82%, 77-87) in the non-responder cohort, and 192 (82%, 77-87) in the ineligible, intolerant, or ineligible and intolerant cohort. Serious adverse events occurred in 12 (6%) patients in the treatment-naive group; 11 (5%) non-responders, and 16 (7%) ineligible, intolerant, or ineligible and intolerant patients; adverse events leading to discontinuation (most commonly reversible increases in alanine or aspartate aminotransferase) occurred in six (3%), two (1%), and two (1%) patients, respectively, with no deaths recorded. Grade 3 or 4 laboratory abnormalities were uncommon, with low incidences of aminotransferase increases during the first 12 weeks with daclatasvir plus asunaprevir and placebo in treatment-naive patients (≤2% each).
Interpretation
Daclatasvir plus asunaprevir provided high sustained virological response rates in treatment-naive, non-responder, and ineligible, intolerant, or ineligible and intolerant patients, and was well tolerated in patients with HCV genotype 1b infection. These results support the use of daclatasvir plus asunaprevir as an all-oral, interferon-free and ribavirin-free treatment option for patients with HCV genotype 1b infection, including those with cirrhosis.
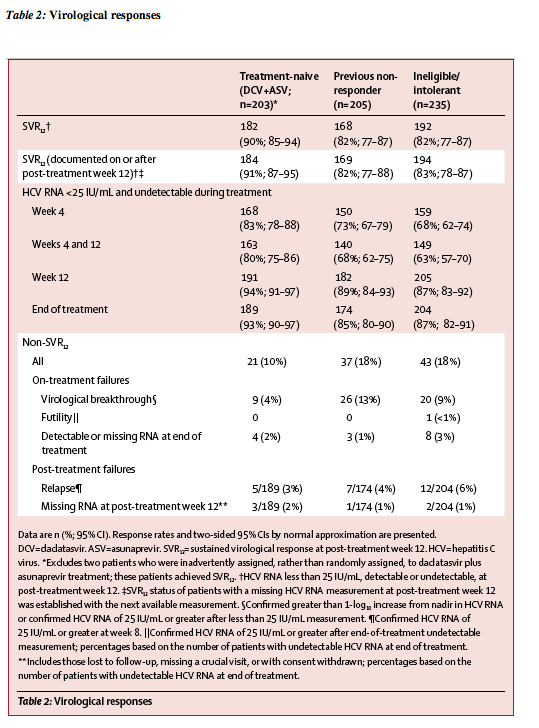
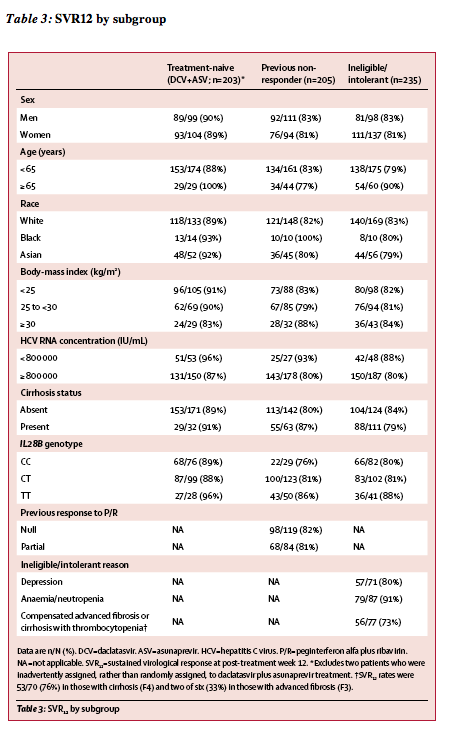
Figure 2: Multivariate logistic regression of SVR12 on baseline covariates
The multivariate analysis included the entire study population, excluding treatment-naive patients who received placebo during the initial 12 weeks, two
treatment-naive patients who were inadvertently assigned, rather than randomly assigned, to daclatasvir plus asunaprevir treatment, and 13 patients who had missing
data (n=630). All parameters included in the analysis were assessed before initiation of study treatment. HCV=hepatitis C virus. SVR12=sustained virological response at post-treatment week 12. *rs12979860 single-nucleotide polymorphism. �The other category included patients who were non-white, non-black, and non-Asian (n=8).
Introduction
Chronic hepatitis C virus (HCV) infection affects an estimated 130-150 million people worldwide, and is a substantial global health problem.1 HCV has seven genotypes; genotype 1 predominates in the USA (about 70% of infections), Europe, north Asia, Australia, and South America, and has historically been the most difficult to cure.2-5 Subtype 1a represents about 60% of genotype 1 infections in the USA, and subtype 1b about 40%.2 In most other countries, subtype 1b is more common than is 1a, and is the most prevalent HCV genotype overall in the large patient populations of east Asia (eg, Japan, China, Korea, Taiwan) and many European countries, such as Italy, Russia, Poland, and Romania.5-7
Approved treatments for HCV genotype 1 infection are limited to combinations containing peginterferon alfa and ribavirin. The newest regimens, which include the direct-acting antivirals simeprevir and sofosbuvir, are substantially more effective than is peginterferon alfa plus ribavirin alone, with response rates of 80% or higher in treatment-naive patients8-10 and lower rates in non-responders to peginterferon alfa plus ribavirin.11 However, because of treatment-limiting adverse events, regimens based on peginterferon alfa plus ribavirin are perceived as poorly tolerated, leading many providers and patients to avoid initiation of therapy. Moreover, many patients cannot tolerate or are medically ineligible for these treatments.12, 13 Therapeutic research has therefore increasingly focused on development of effective and better tolerated all-oral regimens, with several new regimens recently reported.14-18
Daclatasvir is a potent pangenotypic NS5A inhibitor with antiviral activity across HCV genotypes 1-6 in vitro;19 asunaprevir is an NS3 protease inhibitor that is active against genotypes 1, 4, 5, and 6 in vitro.20 Initial clinical assessment of the combination of daclatasvir plus asunaprevir showed high rates of sustained virological response (SVR) in patients with genotype 1b, but reduced efficacy against genotype 1a;21 consequently, subsequent clinical studies have focused on genotype 1b. In a phase 3 Japanese study in genotype 1b, all-oral dual therapy with daclatasvir plus asunaprevir showed high SVR rates and good tolerability in non-responders to peginterferon alfa plus ribavirin (81%), and in patients ineligible for, or intolerant of, peginterferon alfa plus ribavirin (87%).22 In this global study, we assessed the efficacy and safety of daclatasvir plus asunaprevir in treatment-naive and treatment-experienced patients with HCV genotype 1b infection.
Methods
Study design and participants
We did this multinational, phase 3, multicohort study of daclatasvir plus asunaprevir at 116 sites in 18 countries (including in North and South America, Europe, and Asia) between May 11, 2012, and Oct 9, 2013. Eligible patients were aged at least 18 years with genotype 1b infection and HCV RNA of 10 000 IU/mL or greater who met inclusion criteria for one of three cohorts: treatment-naive, previous non-responder to peginterferon alfa plus ribavirin (null or partial response), or ineligible for, intolerant of, or ineligible for and intolerant of peginterferon alfa plus ribavirin (treatment-naive and treatment-experienced). Ineligible or intolerant (or both) patients included those with depression, anaemia or neutropenia, or compensated advanced fibrosis or cirrhosis (F3/F4) with thrombocytopenia. Anaemia was defined as haemoglobin between 85 g/L and less than 120 g/L (women) or less than 130 g/L (men), neutropenia as absolute neutrophils between 0·5 x 109 cells per L and less than 1·5 x 109 cells per L, and thrombocytopenia as platelets between 50 x 109 cells per L and less than 90 x 109 cells per L, at screening or history of these conditions, while receiving peginterferon alfa plus ribavirin, or both (appendix). In all groups, patients with ascites, oesophageal varices, or other evidence of hepatic decompensation were excluded. The protocol was approved by the institutional review board or independent ethics committee at each site. All patients provided written informed consent.
Randomisation and masking
We randomly assigned treatment-naive patients in a 2:1 ratio to receive daclatasvir (one 60 mg tablet once daily) plus asunaprevir (one 100 mg softgel capsule twice daily) or matching placebo for 12 weeks (for comparison of safety and tolerability). The daclatasvir plus asunaprevir group continued open-label treatment to the end of week 24; placebo recipients entered another study and received daclatasvir plus asunaprevir for 24 weeks. Randomisation for the initial 12 week treatment period was done by an interactive voice response system with a computer-generated random allocation sequence, and was stratified by cirrhosis status. Patients and investigator sites were masked to treatment assignment and HCV RNA results to the end of week 12; the sponsor was masked to treatment assignment to the end of week 12. Nonresponders and ineligible, intolerant, or ineligible and intolerant patients received open-label daclatasvir plus asunaprevir for 24 weeks; a placebo group was not included with these patient cohorts because of their generally more advanced liver disease and greater urgency of treatment. Patients receiving daclatasvir plus asunaprevir in all cohorts were followed up for 24 weeks after treatment.
Procedures
We assayed serum HCV RNA concentrations with the High Pure System COBAS TaqMan HCV Test v2·0 (Roche Molecular Systems, Pleasanton, CA, USA); lower limit of quantitation 25 IU/mL) at baseline; on-treatment weeks 1, 2, 4, 6, 8, 12, 16, 20, and 24 (or at early discontinuation); and post-treatment weeks 4, 12, and 24. We assessed the HCV genotype or subtype with the VERSANT HCV genotype 2·0 assay (Siemens, Munich, Germany). We identified the IL28B genotype (rs12979860 single-nucleotide polymorphism) by PCR amplification and sequencing (Applied Biosystems TaqMan assay, Carlsbad, CA, USA). Resistance testing used population sequencing of plasma samples from all patients at baseline and from patients with virological failure and HCV RNA of 1000 IU/mL or more. Safety monitoring was done with adverse event reports, clinical laboratory assessments, vital signs, and physical examinations.
Outcomes
Primary efficacy endpoints were the proportions of treatment-naive patients (receiving daclatasvir plus asunaprevir) and non-responders with SVR (HCV RNA <25 IU/mL, detectable or undetectable) at post-treatment week 12 (SVR12). Secondary efficacy endpoints included SVR12 rates in ineligible, intolerant, or ineligible and intolerant patients, SVR12 by IL28B genotype, proportions with HCV RNA less than 25 IU/mL undetectable at post-treatment week 12, and proportions with HCV RNA less than 25 IU/mL, detectable or undetectable, and less than 25 IU/mL undetectable at weeks 1, 2, 4, 6, 8, 12, and both 4 and 12; at end of treatment; and at post-treatment week 24. Virological breakthrough was defined as confirmed increase from nadir of greater than 1 log10 in HCV RNA or confirmed HCV RNA of 25 IU/mL or greater after a measurement of less than 25 IU/mL, futility as confirmed HCV RNA of 25 IU/mL or greater at week 8, and post-treatment relapse as confirmed HCV RNA of 25 IU/mL or greater after an undetectable end-of-treatment measurement.
Safety endpoints included incidence of adverse events, serious adverse events, discontinuations because of adverse events, anaemia (decrease in haemoglobin to <100 g/L), rash (appendix), and grade 3 or 4 laboratory abnormalities. For the treatment-naive cohort, we assessed differences in rates of grade 3 or 4 laboratory abnormalities between daclatasvir plus asunaprevir and placebo during the first 12 weeks.
Statistical analysis
Target sample sizes were 200 patients in the treatment-naive cohort (receiving daclatasvir plus asunaprevir), 200 in the non-responder cohort, and up to 225 in the ineligible, intolerant, or ineligible and intolerant cohort. These sample sizes would ensure that safety events occurring at a rate of 1·2% or higher (≥1·1% for the ineligible, intolerant, or ineligible and intolerant cohort) would be detected with at least 90% probability; 100 treatment-naive patients receiving placebo would detect with at least 90% probability safety events occurring at a 2·3% rate. With these sample sizes, the width of the 95% CI for the SVR12 rate would be at most 14%.
Efficacy analyses were restricted to patients given daclatasvir plus asunaprevir. In the primary analysis, missing SVR12 data were counted as treatment failures; analysis was also done on the basis of SVR12 documented on or after post-treatment week 12. We computed two-sided 95% CIs for response rates with normal approximations to the binomial distribution. The primary objective for the treatment-naive cohort was to show that the lower bound of the 95% CI for daclatasvir plus asunaprevir was greater than 68% (based on historical results for telaprevir combined with peginterferon alfa plus ribavirin; appendix); the primary objective in other cohorts was to estimate SVR12 rates. Safety analyses were done in patients receiving daclatasvir plus asunaprevir, by cohort, and in the treatment-naive cohort by treatment (daclatasvir plus asunaprevir vs placebo) during the 12 week masked phase (appendix).
This trial is registered with ClinicalTrials.gov, number NCT01581203.
Role of the funding source
The funder, in collaboration with the authors, participated in study design, data collection, data analysis, data interpretation, and writing of the Article. All authors had full access to the data and vouch for the integrity and accuracy of the data reported. The corresponding author had final responsibility for the decision to submit for publication.
Results
This study included 307 treatment-naive patients (205 received daclatasvir plus asunaprevir and 102 received placebo; all randomly assigned patients received the intended treatment), 205 non-responders, and 235 ineligible, intolerant, or ineligible and intolerant patients (143 ineligible, 170 intolerant, and 80 both ineligible and intolerant; figure 1); 190 (93%) treatment-naive patients, 177 (86%) non-responders, and 208 (89%) ineligible, intolerant, or ineligible and intolerant patients completed daclatasvir plus asunaprevir therapy. Table 1 shows baseline demographic and disease characteristics in the patient cohorts. At study entry, cirrhosis, assessed by liver biopsy in 293 patients and by transient elastography in 453 patients with FibroScan (cirrhosis defined as ≥14·6 kPa), was present in 49 (16%) treatment-naive patients; 63 (31%) non-responders; and 111 (47%) ineligible, intolerant, or ineligible and intolerant patients (table 1), which included a subcohort with advanced fibrosis or cirrhosis and thrombocytopenia.
SVR12 rates were 90% (95% CI 85-94) in the treatment-naive cohort, 82% (77-87) in the non-responder cohort, and 82% (77-87) in the ineligible, intolerant, or ineligible and intolerant cohort (91%, 82%, and 83%, respectively, documented on or after post-treatment week 12; table 2). In treatment-naive patients, the lower bound of the 95% CI for the SVR12 rate (85%) exceeded the 68% specified in the primary objective. HCV RNA reductions from baseline were rapid and sustained (appendix); mean decreases at week 2 were 4·8-5·0 log10 IU/mL. HCV RNA was less than 25 IU/mL and undetectable at week 4 in 168 (83%) patients in the treatment-naive cohort, 150 (73%) in the non-responder cohort, and 159 (68%) in the ineligible, intolerant, or ineligible and intolerant cohort, and at the end of treatment in 189 (93%), 174 (85%), and 204 (87%), respectively. SVR12 rates were higher in patients with undetectable (89% [425 of 477]) versus detectable (73% [117 of 161]) HCV RNA at week 4.
We recorded no differences in SVR12 rates based on sex, age, race, body-mass index, or IL28B genotype (table 3). Overall SVR12 rates were similar in patients with cirrhosis (84%) and those without cirrhosis (85%; table 3). In non-responders, SVR12 rates were similar in null responders and partial responders (table 3). Table 3 shows SVR12 rates for subcohorts of ineligible, intolerant, or ineligible and intolerant patients who had depression, anaemia or neutropenia, or advanced fibrosis or cirrhosis with thrombocytopenia in ineligible, intolerant, or ineligible and intolerant patients. In analysis of SVR12 by baseline platelet count in the overall population, 46 of 65 (71%) patients with less than 90 x 109 cells per L achieved SVR12 (appendix). Multivariate regression analysis of baseline factors (figure 2) identified only HCV RNA of 800 000 IU/mL or greater and presence of NS5A resistance-associated variants (at positions L31 or Y93) as negative predictors of SVR12. Notably, SVR12 rates were high in patients with HCV RNA less than 800 000 IU/mL and 800 000 IU/mL or higher (table 3). Baseline NS5A-L31 variants were present in 27 (5%) patients, 11 (41%) of whom achieved SVR12; NS5A-Y93 variants were present in 48 (8%) patients, 18 (38%) of whom achieved SVR12 (appendix).
In patients given daclatasvir plus asunaprevir, 101 of 643 (16%) did not achieve SVR12; on-treatment failures occurred in 13 (6%) patients in the treatment-naive cohort, 29 (14%) in the non-responder cohort, and 29 (12%) in the ineligible, intolerant, or ineligible and intolerant cohort (table 2). NS5A and NS3 variants at aminoacid positions associated with drug resistance were each observed at baseline in about 20% of genotypable isolates for NS5A (n=599) and NS3 (n=634). Other than signature resistance-associated variants at NS5A positions L31 and Y93 (discussed previously) and NS3 position D168, baseline variants in either protein did not seem related to virological outcome (appendix). We detected signature resistance-associated variants at NS5A-L31, NS5A-Y93, NS3-D168, or a combination of two or more in 75 of 596 patients with both NS5A and NS3 baseline sequences, including 27 patients with NS5A-L31, 48 with NS5A-Y93, and three with NS3-D168 variants. Of these 75 patients, 29 (39%) achieved SVR12. 478 of 521 patients (92%) who had both NS5A and NS3 baseline sequences and did not have resistance-associated variants at NS5A-L31, NS5A-Y93, NS3-D168, or any combination thereof achieved SVR12. The most common treatment-emergent variants associated with virological failure were noted at NS5A-L31 (49 of 78 patients with genotypeable isolates), NS5A-Y93 (45 of 78), and NS3-D168 (76 of 83; appendix). We noted these variants together in 61 of 79 (77%) patients with key NS5A and NS3 resistance-associated variants detected at virological failure (including one patient who also had key NS5A and NS3 resistance-associated variants at baseline); the remaining patients had other combinations of NS5A and NS3 variants (appendix).
The most common adverse events (≥10% in any cohort) were headache, fatigue, diarrhoea, nausea, and asthenia (table 4). In patients given daclatasvir plus asunaprevir, 12 of 642 (2%) had anaemia (two of 203 [1%] in treatment-naive cohort, three of 205 [1%] in non-responder cohort, seven of 234 [3%] in ineligible or intolerant [or both]cohort) and 46 of 645 (7%) had rash (16 of 205 [8%], 11 of 205 [5%], and 19 of 235 [8%], respectively). All rash-related events were of mild to moderate intensity, with no treatment discontinuations. Adverse event-related discontinuations were uncommon (table 4). The few discontinuations were most often associated with alanine aminotransferase (ALT) or aspartate aminotransferase (AST) increases (seven patients, five with events more than ten times the upper limit of normal [ULN]; six of seven patients achieved SVR12), which resolved when treatment was discontinued. The appendix summarises all adverse events occurring in 2% or more of patients in any cohort and events causing discontinuation.
Serious adverse events on treatment were reported in 39 patients (6%), with similar incidences across cohorts (table 4; appendix). Investigators deemed four events to be related to daclatasvir, asunaprevir, or both: two atrial fibrillation events in the ineligible, intolerant, or ineligible and intolerant cohort (one each at week 16 and follow-up week 4), and one ALT increased and one event reported as hepatic enzyme increased in the treatment-naive cohort. The event of hepatic enzyme increase occurred at week 24 (after the last dose of daclatasvir plus asunaprevir) in a 26-year-old man with confirmed Gilbert's syndrome (UGT1A1*28 homozygous). Although the patient met prespecified laboratory criteria for potential drug-induced liver injury (ALT ≥five times baseline or nadir and ≥ten times the ULN, and total bilirubin ≥twice the ULN), because of the effect of the history of Gilbert's syndrome on the total bilirubin laboratory criterion, he did not meet the clinical criteria for potential drug-induced liver injury that there be no alternative explanation for either the aminotransferase or bilirubin increases. In this patient, ALT was 690 U/L and total bilirubin was 64 μmol/L at week 24 (46 U/L and 26 μmol/L, respectively, at day 1); direct bilirubin did not increase (7 μmol/L at both times), consistent with Gilbert's syndrome rather than potential drug-induced liver injury being the cause of hyperbilirubinaemia. These increases were asymptomatic with no signs of hepatic decompensation, decreased 9 days after treatment completion, and returned to baseline concentrations within 4 weeks after treatment. The patient achieved SVR12.
Aminotransferase increases of greater than five times the ULN were uncommon, with transient increases in ALT noted in 15 (2%) patients (including the two patients with serious adverse events of ALT increased and hepatic enzyme increased) and transient increases in AST noted in 12 (2%) patients (table 4). The appendix shows median ALT concentrations during the study. Patients with and without cirrhosis had similar frequencies of ALT (1% vs 3%) and AST (1% vs2%) increases greater than five times the ULN. We recorded one haemoglobin reduction to between 70 and 89 g/L; frequencies of other grade 3 or 4 haematological abnormalities were 2% or less in each cohort, apart from a 4% frequency of platelet reductions in ineligible, intolerant, or ineligible and intolerant patients (table 4), which included a subcohort with low platelets at screening (50 x 109 cells per L to <90 x 109 cells per L).
Daclatasvir plus asunaprevir was well tolerated among the subcohorts of ineligible, intolerant, or ineligible and intolerant (or both) patients (appendix), with no cohort-level increase in neuropsychiatric events in the depression subcohort, no increase in measures of bone marrow suppression in the anaemia or neutropenia subcohort, and no worsening of liver synthetic parameters in the advanced fibrosis or cirrhosis subcohort.
During the 12 week masked phase, rates of overall adverse events and grade 3 or 4 laboratory abnormalities were similar in treatment-naive patients receiving daclatasvir plus asunaprevir or placebo; aminotransferase concentrations greater than five times the ULN and total bilirubin increases greater than 2·5 times the ULN were noted in 2% or less of patients in each group (table 4).
Discussion
In this global phase 3 study in patients with genotype 1b infection, including a high proportion with cirrhosis, interferon-free and ribavirin-free all-oral dual therapy with daclatasvir plus asunaprevir provided SVR rates of 82-91% at post-treatment week 12 or later. High SVR12 rates were achieved in treatment-naive patients (91%) and those with high unmet need, such as peginterferon alfa plus ribavirin non-responders (82%) or those ineligible for or intolerant of (or both) peginterferon alfa plus ribavirin (83%; panel). These findings are consistent with those from a phase 3 study in Japanese non-responder or ineligible or intolerant patients with genotype 1b infection (SVR12 rates of 81% and 87%, respectively).22 Notably, in the Japanese study, patients who were ineligible for peginterferon alfa plus ribavirin were treatment-naive, whereas patients in our study were treatment-naive or treatment-experienced. Additionally, compared with our study, the Japanese study included fewer patients with cirrhosis (10% vs 32% [given daclatasvir plus asunaprevir]) and a greater proportion of patients aged 65 years or older (40% vs 21%).
-------------------
Panel
Research in context
Systematic review
We consulted a recent systematic review of hepatitis C virus (HCV) therapies14 and did a PubMed search (up to April 17, 2014) for reports of clinical trials assessing interferon-free treatments for genotype 1 infection (search terms of "HCV" or "hepatitis C", disregarding reports in other genotypes or of interferon-based regimens). We identified several relevant studies.14-18
Interpretation
Treatment for HCV genotype 1 infection is evolving rapidly, with a focus on development of interferon-free and ribavirin-free regimens that provide higher response rates with improved safety and tolerability. In this study, high response rates were achieved in treatment-naive patients and peginterferon alfa plus ribavirin non-responders and ineligible, intolerant, or ineligible and intolerant patients, who have a high unmet medical need. Response rates were similar in patients with and without cirrhosis, and daclatasvir plus asunaprevir was well tolerated. Although interferon-free regimens with higher sustained virological response rates have been reported,14-18 the favourable safety and drug interaction profile of daclatasvir plus asunaprevir supports its potential as a treatment option for patients with genotype 1b infection, including patients who have cirrhosis and those with comorbidities or concomitant medications, or both, and in regions where genotype 1b is prevalent.
------------------
Cirrhosis is associated with decreased SVR rates with peginterferon alfa plus ribavirin alone or in combination with telaprevir, boceprevir, simeprevir, or sofosbuvir.8, 11, 23 In our study, treatment response was similar in patients with or without cirrhosis. The SVR12 rate in patients with baseline platelet counts between 50 x 109 cells per L and less than 90 x 109 cells per L was high (71%), but slightly lower than that in patients without thrombocytopenia (86%), although the sample size was small. This difference might be associated with cirrhosis with portal hypertension, which might result in lower hepatic drug exposure.
Treatment response was generally similar between patients with CC or non-CC IL28B genotypes, which is consistent with previous findings with interferon-free regimens.22,24-27 Sex, age, and race also had no notable effects on treatment outcome. Most virological failures with daclatasvir plus asunaprevir were on-treatment breakthroughs, contrasting with cross-study data for sofosbuvir plus ribavirin in similar patient groups with HCV genotypes 1-3, in which most failures were post-treatment relapses.28 Patients who did not achieve SVR12 with daclatasvir plus asunaprevir (16%) had a higher frequency of baseline NS5A variants at positions L31 and Y93 than did those who achieved SVR12; however, some patients who had these baseline variants still achieved SVR12.
Daclatasvir plus asunaprevir was well tolerated, with low incidences of serious adverse events, adverse events leading to discontinuation, and grade 3 or 4 laboratory abnormalities. The most common adverse events leading to discontinuation were aminotransferase increases; these increases were reversible with no evidence of hepatic decompensation, and six of seven patients who discontinued treatment for this reason achieved SVR12. We recorded no exacerbation of subcohort-specific conditions in ineligible, intolerant, or ineligible and intolerant patients. In treatment-naive patients, frequencies of adverse events and grade 3 or 4 laboratory abnormalities, including aminotransferase and total bilirubin increases, were similar with daclatasvir plus asunaprevir and with placebo during the first 12 weeks of treatment.
This was a global study enrolling a wide range of patients, including those with cirrhosis; the similarity of our results to those from a Japanese study22 is consistent with generalisability of these findings. Limitations of our study include the absence of a placebo group for safety and tolerability comparisons in the non-responder and ineligible, intolerant, or ineligible and intolerant cohorts, and the absence of patients who relapsed on previous peginterferon alfa plus ribavirin therapy. This study also did not assess daclatasvir plus asunaprevir in combination with ribavirin, although results of a study assessing the combination of daclatasvir with another protease inhibitor, simeprevir, showed no substantial differences with or without addition of ribavirin in genotype 1b infection.29
Daclatasvir plus asunaprevir has recently been approved in Japan for the treatment of genotype 1 infection. In the context of other approved therapies, the interferon-free and ribavirin-free regimen of daclatasvir plus asunaprevir provided SVR rates in genotype 1b infection that were similar to, or higher than, those reported for combinations of sofosbuvir or simeprevir with peginterferon alfa plus ribavirin in treatment-naive and treatment-experienced patients.8-11Daclatasvir plus asunaprevir showed reduced frequencies of haematological toxicities and systemic adverse events compared with peginterferon alfa plus ribavirin-based therapies;8, 30, 31 the safety of daclatasvir plus asunaprevir was further shown by comparison with placebo in treatment-naive patients. In this regard, the absence of ribavirin in this combination might be advantageous for patients with an increased risk of ribavirin intolerability, such as those with renal dysfunction, haemoglobinopathies, or vascular disease. Recent publications of other interferon-free regimens have reported SVR rates of 94-99% in treatment-naive and treatment-experienced patients with genotype 1 infection.14-18 On the basis of its favourable safety and drug interaction profile, and high response rates with good tolerability in ineligible, intolerant, or ineligible and intolerant patients with more comorbidities or concomitant medications, or both, daclatasvir plus asunaprevir is a treatment option for this patient population, especially when genotype 1b is highly prevalent. This regimen might not be optimum for genotype 1a infection; however, establishment of HCV subgenotype is common and recommended by guidelines in the USA, European Union, and Asia, because of its importance for therapeutic decision-making.3, 4, 32 Studies are underway to assess addition of a non-nucleoside polymerase inhibitor to daclatasvir plus asunaprevir after promising early results in genotypes 1a and 1b,26 and to assess daclatasvir-containing all-oral combinations in several patient populations with high unmet need.
|
|
| |
| |
|
|
|