| |
Safety, Tolerability, and Pharmacokinetics of Raltegravir After Single and Multiple Doses in Healthy Subjects (2008)
|
| |
| |
Download the PDF here
Clinical Pharmacology & Therapeutics (2008)
M Iwamoto1, LA Wenning1, AS Petry1, M Laethem1, M De Smet1, JT Kost1, SA Merschman1, KM Strohmaier1, S Ramael2, KC Lasseter3, JA Stone1, KM Gottesdiener1 and JA Wagner1
1. 1Departments of Clinical Pharmacology, Clinical Drug Metabolism, Clinical Biostatistics, and Medical Communications, Merck Research Laboratories, a division of Merck & Co., Inc., Whitehouse Station, NJ, USA
2. 2SGS Life Sciences Services, Antwerp, Belgium
3. 3Pharmanet Development Group, Miami, Florida, USA
Abstract
Raltegravir is a novel human immunodeficiency virus-1 integrase inhibitor with potent in vitro activity (95% inhibitory concentration (IC95)=33 nM in 50% human serum). Three double-blind, randomized, placebo-controlled, pharmacokinetic, safety, and tolerability studies were conducted: (1) single-dose escalation study (10-1,600 mg), (2) multiple-dose escalation study (100-800 mg q12 h 10 days), and (3) single-dose female study (400 mg). Raltegravir was rapidly absorbed with a terminal half-life (t1/2) 7-12 h. Approximately 7-14% of raltegravir was excreted unchanged in urine. Area under the curve (AUC)0- was similar between male and female subjects. After multiple-dose administration, steady state was achieved within 2 days; there was little to modest accumulation of raltegravir. Trough levels were >33 nM for dose levels of 100 mg and greater. Raltegravir is generally well tolerated at doses of up to 1,600 mg/day given for up to 10 days and exhibits a pharmacokinetic profile supportive of twice-daily dosing with multiple doses of 100 mg and greater achieving trough levels >33 nM.
The worldwide incidence of human immunodeficiency virus-1 (HIV-1) infection remains considerable, and the number of individuals infected with HIV continues to grow, as does the number of deaths due to AIDS.1 Although advances in the treatment and understanding of the disease have proved beneficial, there still remains an unmet medical need for those infected with HIV-1.1 To date, there are a number of agents available for the treatment of HIV; however, not all infected patients have benefited from them. This is due to the development of viral resistance and medication non-adherence resulting from toxicity.
Reverse transcriptase, protease, and integrase enzymes are key components required for HIV-1 viral replication. Currently available oral medications target reverse transcriptase and protease, interrupting the viral life cycle; however, no integrase inhibitors are presently approved for marketing. HIV-1 integrase catalyzes the stepwise process that results in the integration of the HIV-1 DNA into the genome of the host cell (Figure 1a). This ordered series of reactions includes the assembly of integrase in a stable complex with the viral DNA, the endonucleolytic processing of the viral DNA ends, and the strand transfer or joining of the viral and cellular DNAs (2,3). Integration is required for stable maintenance of the viral genome and also efficient viral gene expression.
Raltegravir (formerly known as MK-0518) is a novel HIV-1 integrase strand transfer inhibitor with potent in vitro activity against HIV-1, exhibiting a 95% inhibitory concentration (IC95) of 33 nM in the presence of 50% human serum (Figure 1b). It is active against a wide range of wild-type and multidrug-resistant HIV-1 clinical isolates and has potent activity against viruses that use CCR5 and/or CXCR4 coreceptors for entry (4,5). Initial results of a 10-day monotherapy proof-of-concept study showed raltegravir to have potent antiretroviral activity as short-term monotherapy (6). In phase II and III studies, which included patients with advanced HIV-1 infection harboring triple-class resistant virus, raltegravir showed a rapid, potent, and sustained antiretroviral effect with dose administration up to 24 weeks and was characterized with a favorable tolerability profile (7,8,9,10)
The metabolism of raltegravir was assessed through in vitro and clinical studies showing that raltegravir is primarily cleared by metabolism with a small component ( 9% of the administered dose) of elimination via renal excretion (11) Metabolism was primarily by glucuronidation mediated by the uridine diphosphate glucuronosyl transferase 1A1 isozyme.
This report describes the results of three phase-I studies conducted in healthy subjects to investigate the single- and multiple-dose pharmacokinetics of raltegravir. These studies also assessed the potential pharmacokinetic differences between male and female subjects.
Results
Pharmacokinetics
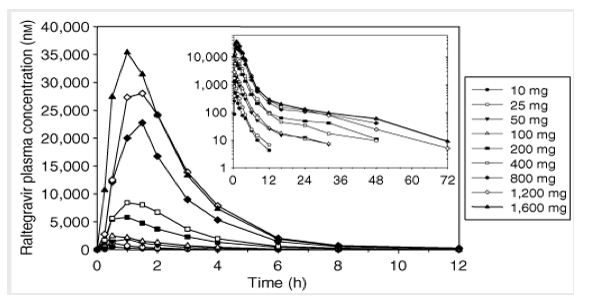
Study I: single-dose escalation study. The plasma raltegravir concentration profiles and the principal raltegravir pharmacokinetic parameters are summarized in Figure 2 and Table 1. Raltegravir appears to be rapidly absorbed, with median values of time to peak plasma concentration (Tmax) in the fasted state ranging from 0.5 to 1.3 h. Raltegravir concentrations declined from the mean peak plasma concentration (Cmax) in a biphasic manner, with an apparent half-life of the initial ( ) phase of approximately 1 h and an apparent half-life of the terminal ( ) phase of approximately 7-12 h. Increases in area under the curve (AUC) and plasma concentration at 12 h post-dose (C12h) were approximately proportional to dose over the dose range of 10 mg to 1200 mg. Increases in Cmax were slightly less than dose-proportional, and Tmax values were slightly longer at higher doses than lower doses. Approximately 7-14% of the oral raltegravir dose was excreted unchanged in urine, and the renal clearance was approximately 42-78 ml/min. After single-dose administration of raltegravir at dose levels of 200 mg and higher, the geometric mean C12 h exceeded 33 nM (in vitro IC95 in 50% human serum).
Figure 1. Mean raltegravir plasma concentrations (nM) versus time (h) following single-dose administration of 10-1,600 mg raltegravir (lactose formulation) in the fasted state to young, healthy male subjects (inset: semilog scale).
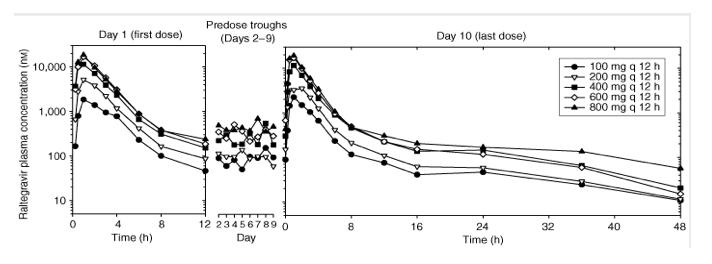
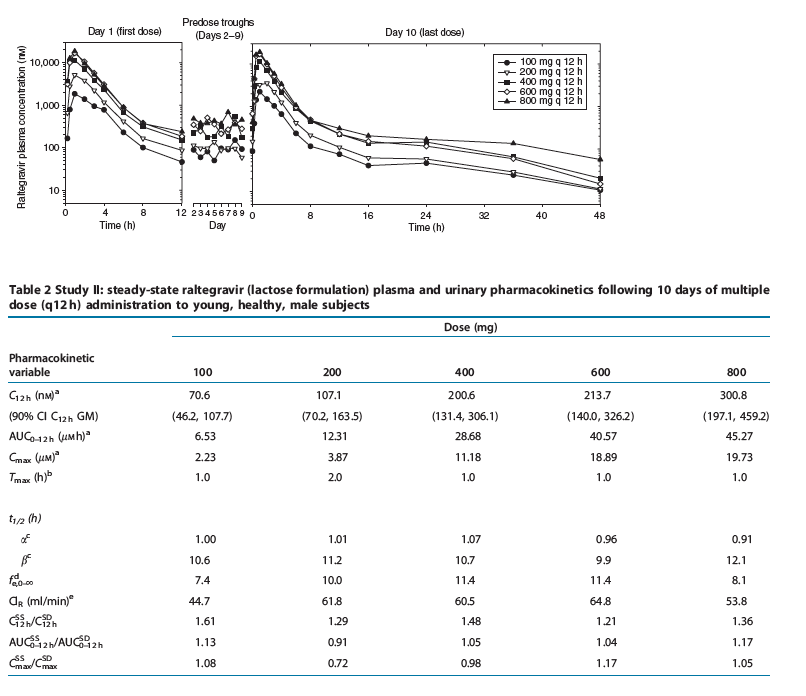
Study II: multiple-dose escalation study. The plasma raltegravir concentration profiles and the principal raltegravir pharmacokinetic parameters are summarized in Figure 3 and Table 2. After multiple-dose administration of raltegravir, steady state appears to have been achieved within 2 days of dosing (Figure 3). The C12 h geometric mean on day 10 exceeded 33 nM for dose levels of 100 mg and higher. AUC0-12 h and Cmax increase approximately dose proportionally, whereas C12 h appears to increase moderately less than dose proportionally. The apparent terminal elimination half-lives following the final dose were approximately 1 h for the initial ( ) phase and approximately 10-12 h for the terminal ( ) phase. The average accumulation ratios (steady state versus single dose) for AUC0-12 h and Cmax, over the dose range studied, ranged from approximately 0.7 to 1.2, indicating little, if any, accumulation in these parameters with every 12 h (q12 h) dosing. The average accumulation ratio for C12 h ranged from approximately 1.2 to 1.6, indicating modest accumulation. Approximately 8-11% of the oral raltegravir dose was excreted unchanged in urine during a steady-state dosing interval, with renal clearance values of approximately 54-65 ml/min.
Figure 3. Mean raltegravir plasma concentrations (nM) versus time (h) and assessment of steady state following multiple-dose administration of 100-800 mg raltegravir (lactose formulation) q12 h in the fasted state to young, healthy male subjects (semilog scale).
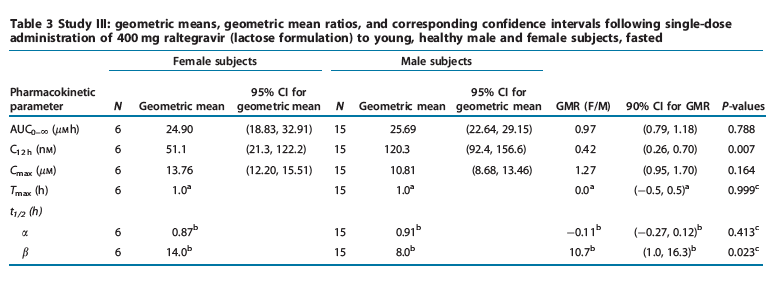
Study III: single-dose female study. The principal raltegravir pharmacokinetic parameters are summarized in Table 3. Overall exposure to raltegravir (assessed through comparison of AUC0- values) was similar in both female and male subjects. C12 h values were modestly lower in female subjects (average 58% lower) than male subjects. The difference observed in Cmax values demonstrated an average increase of 27% in female compared to male subjects. Initial half-lives were similar in both female and male subjects; however, a modestly longer terminal half-life was observed in female subjects (harmonic mean (jackknife SD) t1/2 was equal to 14.0 (9.7) h in female subjects and 8.0 (3.6) h in male subjects).
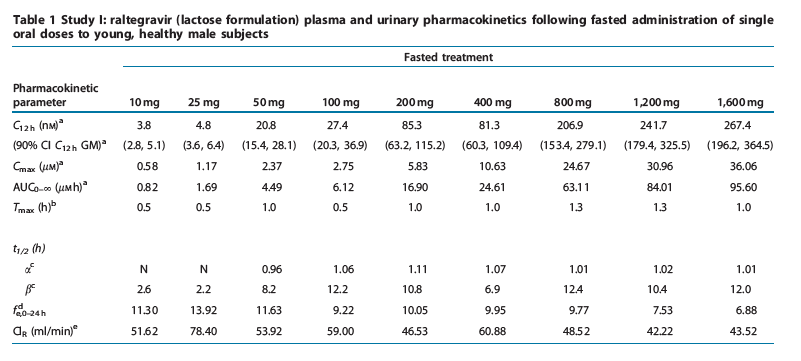
Figure 3 Mean raltegravir plasma concentrations (nM) versus time (h) and assessment of steady state following multiple-dose administration of 100-800mg
raltegravir (lactose formulation) q12 h in the fasted state to young, healthy male subjects (semilog scale).
Safety and tolerability
Raltegravir was generally well tolerated. No serious clinical or laboratory adverse experiences were reported and no subject discontinued because of an adverse experience. Of the 93 non-serious clinical adverse experiences reported by 40 subjects, 25 were considered by the investigator to be possibly related to study drug. The most common drug-related adverse experiences were headache and fatigue. All adverse experiences reported were transient and mild to moderate in intensity. Laboratory adverse experiences were not observed, and neither were consistent treatment-related changes in laboratory, vital signs, or electrocardiogram safety parameters.
Discussion
The combined data from the three clinical studies assessed the pharmacokinetics, safety, and tolerability of raltegravir administered as single and multiple doses to healthy subjects. In the initial single- and multiple-dose escalation studies, raltegravir was absorbed rapidly, with median Tmax values in the fasted state of 1 h; plasma concentrations decreased from Cmax in a biphasic manner, with a half-life of approximately 1 h for the initial ( ) phase and an apparent half-life of approximately 7-12 h for the terminal ( ) phase. With multiple-dose administration, steady state was achieved rapidly, within 2 days of dosing, and there was little accumulation. Over the dose range studied, raltegravir pharmacokinetics were approximately to slightly less than dose proportional.
Raltegravir is an agent in a new class of antiretrovirals, and there are insufficient clinical data defining the target pharmacokinetic parameter. For other classes of antiretroviral agents, there is a reasonable but imperfect association of efficacy with doses that achieve Ctrough values that exceed the effective viral inhibitory concentration (i.e., protein-adjusted IC95) in the HIV spread assay (12,13,14,15). In view of the IC95 value of raltegravir in the HIV spread assay (50% human serum), the target trough level is 33 nM; however, it is unknown whether efficacy would be observed with raltegravir doses that achieve trough levels lower than this IC95 target. After multiple-dose administration, the geometric mean plasma concentration at 12 h post-dose (C12 h) exceeded 33 nM at doses of 100 mg and higher. On the basis of these findings, pharmacokinetic data for raltegravir are supportive of twice daily administration. Efficacy studies in individuals infected with HIV-1 demonstrated that doses from 100 to 600 mg administered twice daily were associated with potent antiviral response (6,7,8,9,10)
Approximately 7-14% of the raltegravir dose was excreted unchanged in urine, and the renal clearance was approximately 42-78 ml/min. As determined in vitro, the fraction of raltegravir that is unbound to plasma proteins in humans is 17%, and therefore assuming a typical glomerular filtration rate of 120 ml/min, a renal clearance value of approximately 20 ml/min would be anticipated for raltegravir based on filtration alone. The observed renal clearance values in this study were somewhat higher than this value, implying that raltegravir may be actively excreted into urine. However, based on the low percent of dose excreted unchanged into urine, renal clearance probably plays a fairly minor role in the overall elimination of raltegravir. The amount of raltegravir excreted unchanged in urine is similar to that characterized in the human absorption, distribution, metabolism, and elimination study, where 9% of the total dose was excreted in the urine as parent compound (11). In vitro and in vivo characterization of the metabolism have shown that raltegravir is primarily eliminated by metabolism via glucuronidation with uridine diphosphate glucuronosyl transferase 1A1.
Raltegravir pharmacokinetics were evaluated in female and male subjects. Overall exposure to raltegravir, as assessed by AUC, was similar between genders. There was no clinically meaningful difference in Cmax values. Female subjects had modestly lower C12h values and a longer apparent terminal t1/2 than male subjects. Raltegravir data obtained from male subjects in other pharmacokinetic studies (e.g., data from study I) showed some interstudy variability in pharmacokinetic parameters. The magnitude of the difference observed in C12 h between male and female subjects is thus dependent upon the choice of comparator group, with pharmacokinetic comparisons from previous studies in male subjects resulting in smaller differences. Because of the longer apparent terminal t1/2 seen in female subjects, differences may cease to exist on multiple dosing with potentially more accumulation in C12 h. Taken together, these results supported inclusion of women in phase II and III efficacy studies without a dose adjustment.
The formulation of raltegravir used in this study (the lactose formulation) was a probe formulation used in the initial clinical studies. The formulation in phase II and III studies differed; however, the general pharmacokinetic behavior of lactose formulation provided important initial information with regard to the development of the clinical program.
In these studies, raltegravir was generally well tolerated when given as single doses, ranging from 10 to 1,600 mg, and when given twice daily (q12 h) for 10 days at doses ranging from 100 to 800 mg. There were no qualitative differences in the incidence of any adverse experience from placebo and no increase in the incidence of adverse experiences (overall or of any specific type) with increasing dose. Many of the presently available anti-HIV drugs, including the nucleoside and non-nucleoside reverse transcriptase inhibitors and protease inhibitors, have toxicity and tolerability issues.16 Raltegravir, an HIV integrase inhibitor, is in a new class of antiretrovirals, which has demonstrated a clean safety profile in this study at doses of up to 1,600 mg/day for up to 10 days and may not have the same associated toxicity and tolerability issues as agents marketed at present. The long-term safety and tolerability profile of raltegravir is the subject of ongoing studies.
In summary, raltegravir is generally well tolerated in young, healthy male and female subjects and exhibits a pharmacokinetic profile supportive of twice-daily dosing, with multiple doses of 100 mg and higher achieving trough levels of >33 nM.
Methods
Subjects. A total of 81 healthy male (N=73) and female (N=8) volunteers were enrolled in the three studies. All subjects were non-smokers with a mean age of 37 years (range: 18-50 years) for male subjects and 37 years (range: 22-45 years) for female subjects and weighed within 20% of ideal body weight with a mean weight of 81 kg (range: 61-107 kg) for male subjects and 66 kg (range: 51-87 kg) for female subjects. Subjects were HIV-negative and in good general health according to routine medical history, physical examination, vital signs, and laboratory data.
Every subject gave written informed consent to participate in the studies. The protocols were approved by the institutional review boards of the respective study centers. The protocols were conducted in accordance with the guidelines on good clinical practice and with ethical standards for human experimentation established by the Declaration of Helsinki Principles.
Study designs. Study I: single-dose escalation study. Study I was a double-blind, randomized, placebo-controlled, alternating panel, multiple-period, rising single-dose study in 24 young, healthy male subjects. In panel A, subjects were administered 10, 50, 200, and 800 mg of raltegravir or placebo; in panel B, subjects were administered 25, 100, 400, and 1,200 mg of raltegravir or placebo. In each panel, six subjects received raltegravir (lactose formulation) and two subjects received placebo. Subjects were given single oral doses of raltegravir or placebo in the fasted state; two different subjects received placebo for each dose level in a randomized balanced manner. Doses were administered in an alternating-panel manner proceeding from the lowest dose to the highest. Following completion of dosing for panels A and B, a third group of eight subjects (panel E) received a single oral dose of 1,600 mg raltegravir or placebo in the fasted state. Subjects in panel E also participated in panel E of the multiple-dose escalation study (see below).
Study II: multiple-dose escalation study. Study II was a double-blind, randomized, placebo-controlled, serial-panel, rising multiple-dose study in 40 young, healthy male subjects. Five panels (panels A-E) of eight subjects each received 100, 200, 400, 600, or 800 mg of raltegravir (lactose formulation) or placebo every 12 h (q12 h) for 10 consecutive days. On days 1 and 10, the morning dose of study drug was administered in the fasted state. Two of the eight subjects in each of the panels received placebo instead of raltegravir. Panel E participants also participated in panel E of study I. Dose escalation for study I up to 1,200 mg and study II up to 600 mg occurred before dosing of 1,600 mg in study I and 800 mg q12 h in study II. Owing to the temporal association of the conduct of the two studies and the collective favorable safety and pharmacokinetic data collected in both studies, the same subjects (those in panel E) were enrolled for dosing of 1,600 mg in study I and 800 mg q12 h in study II.
Study III: single-dose female study. Study III was a double-blind, randomized, placebo-controlled, single-dose study in young, healthy female and male subjects. Eight female subjects were randomly assigned to receive a single oral dose of either 400 mg raltegravir (lactose formulation) (N=6) or placebo (N=2) in the fasted state. Fifteen male subjects received 400 mg single oral dose of the raltegravir in an open-label manner in the fasted state. The male subjects were also involved in a separate pharmacokinetic study that required more subjects than the comparative female cohort. This resulted in an unequal number of male and female subjects in the comparison. On the basis of previous safety data collected in males, no placebo controls were used in the male cohort.
Pharmacokinetic assessments. Plasma and urine samples were collected and analyzed for raltegravir concentrations. In study I, blood samples were drawn and plasma was collected predose and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 32, 48, and 72 h after dosing in each treatment period. Urine for raltegravir assay was collected from 0 to 24 h after dosing in each treatment period. In study II, plasma for raltegravir assay was collected predose and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, and 12 h after the morning dose on days 1 and 10. On day 10, plasma was also collected at 16, 24, 36, 48, and 72 h after the morning dose. Urine for raltegravir assay was collected before the morning dose on day 1 and from 0 to 12 h after the morning dose on day 10. In study III, plasma was collected predose and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 32, 48, and 72 h after dosing.
The analytical method for the determination of raltegravir in human plasma involved isolation, via 96-well liquid-liquid extraction, of the analyte and internal standard from plasma, followed by reverse-phase high-pressure liquid chromatography with tandem mass spectrometry in the positive ionization mode, using an atmospheric pressure chemical ionization interface (Applied Biosystems API 4000, Foster City, CA). The analytical method for the determination of raltegravir in human urine involved sample dilution and direct injection onto the high-pressure liquid chromatography-tandem mass spectrometry system. The lower limit of quantitation for the plasma assay was 2 ng/ml (4.5 nM), and the linear calibration range was 2-1,000 ng/ml. The lower limit of quantitation for the urine assay was 0.25 g/ml, and the linear calibration range was 0.25-25 g/ml. The column used was an Ace C18 (3.0 50 mm, 3 m). The mobile phase consisted of 42.5/57.5 (v/v%) 0.1 mM ethylenediaminetetraacetic acid in 0.1% formic acid/methanol, and the flow rate was 0.5 ml/min. The mass transitions were 445-109 (m/z) for raltegravir and 451-367 (m/z) for internal standard. Two sets of low-, medium-, and high-quality control samples were evaluated with each run of clinical samples. Overall for the individual studies, the interday accuracy of the plasma quality control samples was within 5% of nominal and the interday precision was less than 8%. The urine quality control samples interday accuracy was within 8.4% of nominal and the interday precision was 4.2% or lower.
The distribution and elimination phases of each single dose and day 10 multiple-dose plasma concentration profile were fit to a biexponential equation (concentration=Ae- t+Be- t), using the Gauss-Newton (Levenberg and Hartley) minimization method and a weighting of 1/(predicted concentration)2. Onset of the -phase was determined by inspection. Half-lives (t1/2) for each phase were calculated as the quotient of ln(2) and or . For all subjects at single doses of 10 and 25 mg fasted and isolated subjects at single doses of 400 mg fasted, two log-linear phases were not apparent in their plasma concentration profiles. For these subjects, a monoexponential equation was used instead (concentration=Be- t). For single-dose data, AUC to the last time point with a detectable plasma concentration (AUC0-last) was calculated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations. AUC0- was estimated as the sum of AUC0-last and the extrapolated area given by the quotient of the last measured concentration and . For multiple-dose data, AUC0-12 h was calculated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations. For both single- and multiple-dose data, Cmax and Tmax were obtained by inspection of the plasma concentration data. C12 h and C24 h values were calculated from the plasma concentrations determined for the nominal sampling times at 12 and 24 h post-dose, respectively. For multiple-dose data, the accumulation ratio for AUC was calculated as the ratio of AUC0-12 h for the last dose (day 10) versus the first dose (day 1). Accumulation ratios for Cmax and C12 h were calculated in an analogous manner.
Raltegravir urine concentrations, urine volumes from individual collection intervals, and nominal times of collection intervals were used to calculate urinary pharmacokinetic parameters. The amount of raltegravir excreted unchanged in urine in each collection interval was determined by the product of the urine concentration and the urine volume. The percent of the raltegravir dose that was excreted unchanged in urine over the collection interval (fe,0- ) was determined by the quotient of the sum of raltegravir collected over all collection intervals and the dose administered, with the result multiplied by 100. Following each dose, AUC0- was calculated, where represents the nominal stop time of the final urine collection interval (24 h for single dose data and 12 h for multiple dose data). Renal clearance (ClR) was determined as the quotient of fe,0- and AUC0- .
Safety and tolerability. Safety and tolerability were assessed in each study by clinical evaluation (including physical examinations, vital signs, and 12-lead electrocardiograms) and laboratory measurements (including hematology, serum chemistry, and urinalysis). Adverse experiences were monitored throughout the study. Investigators evaluated all clinical adverse experiences in terms of intensity (mild, moderate, or severe), duration, severity, outcome, and relationship to study drug.
Statistical analyses. Study I: Raltegravir pharmacokinetic parameters (AUC0- , Cmax, and C12 h) were analyzed using an analysis of variance model appropriate for the alternating panel rising dose design.
Study II: Raltegravir pharmacokinetic parameters were analyzed using an analysis of variance model appropriate for the study design.
Study III: A two-sided 90% confidence interval was constructed for the geometric mean ratio (females/males) of raltegravir pharmacokinetic parameter values using a two-sample t-test.
|
|
| |
| |
|
|
|