 |
|
|
| |
The 15th International Workshop on Clinical
Pharmacology of HIV and Hepatitis Therapy
|
| |
| |
Courtney V. Fletcher, Pharm.D.
Dean and Professor
College of Pharmacy
University of Nebraska Medical Center
Jennifer J. Kiser, Pharm.D.
Assistant Professor
School of Pharmacy
University of Colorado at Denver
The 15th International Workshop on Clinical Pharmacology of HIV & Hepatitis Therapy was held in Washington, DC, from May 19-21, 2014. This year, two separate meetings were integrated into one and marks the beginning of what we believe is the premier meeting on the clinical pharmacology of HIV and hepatitis therapy. In this report we will highlight abstracts focused on pharmacologic issues that are of broad interest or might benefit from some expert clarification. Abstracts will be discussed in two broad categories: (i) the clinical pharmacology of therapy for HIV infection, and (ii) the clinical pharmacology of therapy for hepatitis infection. This report does not cover all plenaries and data presented at the Workshop. For more information and to view presentations from the meeting please visit http://www.infectiousdiseasesonline.com/event/workshop/15th-int-wksp-clin-pharmacology-hiv-hepatitis-therapy/.
NATAP coverage: 15th International Workshop on Clinical Pharmacology of HIV and Hepatitis Therapy May 19- 21, 2014, Washington, DC
Abbreviations
%CV, percent coefficient of variation
3TC, lamivudine
ARV, antiretroviral drug
ART, antiretroviral drug therapy
AUC, area under the concentration-time curve
BCRP, breast cancer resistance protein
C12, drug concentration at 12 hours post dose
Cmax, maximum drug concentration
CSF, cerebrospinal fluid
CT, cervical tissue
Ctrough, concentration immediately before the next dose
CYP, cytochrome P450 drug metabolizing enzymes
DBS, dried blood spot
DRV, darunavir
EC90, 90% effective concentration
EFV, efavirenz
ETR, etravirine
FDC, fixed dose combination
FTC, emtricitabine
GMR, geometric means ratio
HCV, Hepatitis C virus
IC50, concentration of drug required to inhibit viral replication in vitro by 50%
IC95, concentration of drug required to inhibit viral replication in vitro by 95%
LDV, ledipasvir
LPV, lopinavir
MVC, maraviroc
NRTI, nucleoside reverse transcriptase inhibitor
NNRTI, non-nucleoside reverse transcriptase inhibitor
OAT, organic anion transporter
OATP, organic anion-transporting polypeptide
OCT, organic cation transporter
PBMCs, peripheral blood mononuclear cells
PD, pharmacodynamic
P-gp, p-glycoprotein
PK, pharmacokinetic
PI, inhibitor of viral protease
PrEP, pre-exposure prophylaxis
QD, once daily
r or RTV, ritonavir
RAL, raltegravir
RBC, red blood cells
RBV, ribavirin
RPV, rilpivirine
RT, rectal tissue
SOF, sofosbuvir
TDF, tenofovir disoproxil fumarate
TFV, tenofovir
TFV-DP, tenofovir diphosphate
VL, viral load as measured by HIV-RNA
VT, vaginal tissue
The Clinical Pharmacology of Therapy for HIV Infection.
Courtney V. Fletcher, Pharm.D.
I. Studies of Dose Reduction.
1. Efavirenz (EFV) pharmacokinetic (PK) and viral load data from the ENCORE1 study (abstract O_01).
ENCORE1 was a randomized, placebo controlled trial that compared an EFV dose of 400 mg QD with the usual 600 mg QD (both with TDF/FTC) in 630 ARV-naïve persons in 13 countries. The results were published Feb 10, 2014 in Lancet. Key findings were: no difference in the proportion of subjects at wk 48 with viral load <200 cpm (400 mg, 94.1% vs 600 mg, 92.2%); no difference in the overall proportions of participants reporting adverse events or the severity of adverse events; no statistical difference in proportions with CNS adverse effects; and fewer patients in the 400 mg arm discontinued therapy from drug-related adverse events (400mg, n=6, 2% vs. 600mg, n=18, 6%).
Abstract O_01 reported the results of a population PK study in 606 participants of the ENCORE1 study. These investigators had previously reported the results of an intensive PK study at CROI 2014 (abstract 510) that showed, as expected, the EFV concentrations with the 400 mg dose were lower than those with the 600 mg dose: for example, the C12 was 1500 ng/mL in the 400 mg group compared with 2050 ng/mL in the 600 mg group. In the present analysis of 606 subjects with PK data and 593 subjects with wk 48 viral load data, investigators found that EFV concentrations were strongly associated with the odds of achieving a viral load at wk 48 and <200 copies/mL (odds ratio, 3.3, 2.0-7.0, p=0.0001). The proportion of patients with a C12 hour concentration <1000 ng/mL who had a viral load ≥200 copies/mL was 21% (4/19) compared with 2% (2/574) in those with C12 values >1000 ng/mL. Of the 19 with a C12 <1000 ng/mL, 14 were receiving 400 mg QD and five 600 mg QD.
Some knowns.
The PK data are consistent with expectations: when the dose of EFV is reduced by 30%, EFV concentrations are reduced similarly. EFV concentrations are associated with virologic response. A mid dose interval EFV concentration (e.g. C12) of ≥1000 ng/mL has frequently been found to be associated with an increased probability of viral load suppression, and this was found in ENCORE1. A higher proportion of patients will achieve this threshold who receive an EFV dose of 600 mg QD than do who receive 400 mg QD.
Some unknowns.
First, we don't know whether an EFV 400 mg QD dose could be equivalent to the 600 mg dose in achieving suppression to <200 cpm but not equivalent for <50 cpm. Second, 4 patients in ENCORE1 had both C12 <1000 ng/mL and viral load ≥200 copies/mL: 1 and 3 were receiving 400mg and 600mg, respectively. This observation is not consistent with other ENCORE1 data, and though we can't dismiss it completely, these numbers are just too small to draw any conclusions and there are no objective data on patient medication adherence.
Population PK-PD Analysis of 400 mg vs. 600 mg Efavirenz Once Daily in Treatment-Naïve HIV Patients at 48 Weeks: Results of the ENCORE1 Study - (05/21/14)
2. A reduced darunavir (DRV) dose is effective at maintaining HIV suppression (abstract O_02).
100 HIV-infected patients with VL <50 cpm receiving DRV/r (800/100 once daily) and 2 NRTIs were randomized to continue on this regimen or to have their DRV dose reduced to 600/100 QD (DRV600 Study). After 48 weeks, 2 patients in the DRV 800mg arm and 3 in the DRV 600mg arm developed virologic failure (two consecutive HIV RNA >50 cpm). One patient in each arm was lost to follow-up and one in the DRV 600mg arm died from liver cirrhosis and septic shock. The intent to treat analysis thus found a 94% rate of maintenance of VL suppression with the DRV 800mg dose and a 90% rate with the 600mg dose (difference, -4%; 95% CI, -12.9, 4.9, p=0.46). There was no difference (p=0.78) in DRV trough concentrations over the 48 weeks, averaging around 2000 ng/mL in both arms. Similarly, CD4 cell counts remained stable in both arms and there was no difference in adverse events. These data provide a basis to suggest that in stable patients who have achieved and maintained a VL reduction to <50 cpm for >3 months, that a reduced dose of DRV to 600 mg once daily, given with 100mg of RTV, is equivalent to the standard dose of 800/100 mg once daily. A cost analysis of drug regimen costs only indicated a saving of 1000 euros (≅ $1360) per patient per year with the 600mg dose.
Reduced Darunavir Dose Is as Effective in Maintaining HIV Suppression as the Standard Dose in Virologically Suppressed HIV-Infected Patients. The DRV600 Study - (05/21/14)
And, some final thoughts about dose reduction strategies.
I believe the ENCORE1 data and the DRV600 Study provide compelling evidence to investigate further whether the starting dose of EFV can be reduced and whether the maintenance dose of DRV can be reduced. But, lets proceed very carefully to evaluate these and other reduced dose strategies. The losers in an ill-conceived race to lower doses are the patients - virologic failure and resistance have real consequences, and the cost of treating these complications, I believe, will far exceed any cost savings simply from a dose reduction.
II. Compartments.
1. CSF concentrations of DRV are similar in patients receiving DRV/RTV 600/100 once daily and 800/100 once daily (abstract P_46). In a companion abstract to the DRV600 study discussed above, CSF concentrations of DRV and CSF viral load were measured in 16 patients, 8 each receiving DRV/RTV 600/100 and 800/100. In the DRV-600 group, the median CSF concentrations were 17.1 ng/mL. In the DRV-800 group, the median CSF concentration was 13.2 ng/mL. In both groups, the CSF sample was obtained a median of 26h after the last dose. All patients had plasma VL <40 cpm, and 14 had CSF VL <40 cpm. Two patients had CSF VL >40 cpm, one in each DRV dose group. These patients had the lowest CSF concentrations of DRV, 5.5 ng/mL and 3.5 ng/mL. These data provide support that in the CSF compartment, the concentrations and viral load suppression achieved with DRV-600 appear equivalent to those with DRV-800. Though the number of patients with CSF VL >40cpm was very small, the finding of a measurable VL and the lowest CSF concentrations of DRV argues for continued exploration of an association between low CSF concentrations and CSF viremia.
DRV CONCENTRATIONS AND VIRAL LOAD IN CSF OF PATIENTS ON DRV/r 600/100 OR 800/100 mg ONCE DAILY, PLUS TWO NRTI - (05/23/14)
2. Vaginal, cervical and rectal tissue concentrations of maraviroc (MVC) and raltegravir (RAL) in women (abstract O_08). 48 women received single doses of MVC (150mg, 300mg and 600 mg) and RAL (200mg, 400mg and 800mg). Plasma and vaginal (VT), cervical (CT) and rectal (RT) tissue samples were collected over 48hours after the dose. For both drugs, distribution into VT and CT was rapid and concentrations in VT and CT were very similar. MVC appeared in RT rapidly, but RAL was somewhat delayed with a maximum observed concentration occurring at 24 hours. RT concentrations were 10X higher for MVC and 25x higher for RAL than in CT/VT tissue. Plasma concentrations did not predict RT concentrations. While no conclusions about potential effectiveness for PrEP can be drawn, these data do provide a PK basis for further studies of MVC. The high RT concentrations observed with RAL may also provide a basis for further studies to prevent rectal transmission. However, a recently published PK study (http://www.ncbi.nlm.nih.gov/pubmed/24687503) examined RAL concentrations in CT in healthy women receiving 400 mg twice daily over 22 days and found concentrations were not higher than plasma and that ≅ 10% of all CT trough concentrations were less than the IC95. These data raise doubt as to whether RAL is a viable agent to prevent vaginal/cervical HIV transmission.
Mucosal Tissue Pharmacokinetics of Maraviroc and Raltegravir in Women: Implications for Chemoprophylaxis [PrEP, Mucosal Tissue Compartments PK] - (05/21/14)
3. Measurement of TFV-diphosphate in red blood cells (RBCs) as a measure of PrEP adherence (abstract PP_05). It is now very clear that adherence is a significant stumbling block to achieving high levels of PrEP effectiveness. Steve Becker, M.D. with the Gates Foundation gave a superb presentation at this meeting on HIV prevention and socio-behavioral pharmacology - that is, factors that motivate drug taking behavior. In his presentation, Steve commented that the VOICE Trial, which enrolled 5029 women and failed because of ≅ 20% adherence, cost approximately $120 million. I've commented before that we urgently need tools that can easily and inexpensively be used in real-time to identify non-adherence and a support structure for study participants and patients that can implement interventions to improve adherence. Abstract PP_05 reports a novel approach to measuring adherence by measuring TFV-diphosphate (TFV-DP) in red blood cells (RBCs) rather than in the usual PBMCs. The advantage of using RBCs is that the half-life of TFV-DP in RBCs is ≅ 17 days vs. 3-4 days in PBMCs. Thus, you get a better picture of long-term medication adherence. In a study of 39 participants (21 HIV-infected, 18 HIV-negative), these investigators found the half-life of TFV-DP was 18.5 days and there was a low between patient variability; neither sex, race or body weight significantly influenced the half-life. These data confirm the long half-life of TFV-DP in RBCs and show its universal applicability since it is not affected by various patient characteristics, and provide a solid basis for investigation in a clinical trial.
4. The estimated onset and duration of PrEP activity for daily TDF/FTC using EC90 data from iPrEx (abstract O_10). In 2014, the CDC published clinical practice guidelines for PrEP (see http://www.cdc.gov/hiv/pdf/PrEPguidelines2014.pdf). With regard to when protection is achieved after starting oral TDF/FTC, the guidelines conclude: "The time from initiation of daily oral doses of TDF/FTC to maximal protection against HIV infection is unknown." This is a question that both health care providers and patients need answered. In one of the most important abstracts presented at this meeting, Peter Anderson and colleagues provided some much needed information. These investigators used PBMC and rectal mononuclear cell intracellular concentrations from intensively studied HIV-negative volunteers and evaluated the time to reach the EC90 (onset of protection) and once steady-state concentrations were achieved, how long concentrations remained above the EC90 after therapy was discontinued (duration of protection). The EC90 is the intracellular TFV-DP concentration associated with 90% protection of HIV transmission in MSM in the iPrEX study (see http://www.ncbi.nlm.nih.gov/pubmed/?term=22972843 ). For the onset of protection, the analysis found that after 7 days of once daily TDF/FTC dosing, 88% of subjects (95%CI, 65-96) would achieve the EC90. For the duration of protection, 86% of subjects (95%CI, 63%-95%) still had concentrations above the EC90 two days after stopping therapy. The iPrEX regression formula was then used to estimate the degree of HIV risk reduction. The inferred risk reductions for onset of protection were 95% (95%CI, 92%-98%) after 3 days of dosing and 99% (95%CI, 98%-100%) after 7 days of dosing. The inferred risk of protection for the duration of protection was >95% for 6 days after stopping therapy.
These data provide some guidance for the implementation of PrEP: that, though some level of protection will be seen after 3 days of TDF/FTC therapy, 7 days should be recommended before engaging in sexual activity; and that there will be a loss of protection after skipping therapy for 2 days though some level of protection may last as long as 6 days after stopping therapy. It must be stressed that these data are derived from MSM. The obvious, next question is what about heterosexual men and women and injection drug users? To my knowledge, no data such as these derived from MSM are available. That said, I am not aware of any reason to suspect that the onset of protection will be shorter. For all populations taking PrEP, guidance on the duration of protection with a drug holiday or after drug discontinuation is a more complex. While the data from this present analysis in MSM indicate some protection may last as long as 6 days, the data and recommendations on the duration that post exposure prophylaxis (PEP) should be continued are also relevant. Specifically, that in vitro, animal, and occupational studies showed 4 weeks of PEP appeared protective (see: http://www.cdc.gov/hiv/risk/other/occupational.html and http://www.jstor.org/stable/10.1086/672271
Until other data are available, I think it is wise to recommend that TDF/FTC therapy be continued for 4 weeks after the last (potential) exposure. Data on the onset of protection from heterosexual man and women and injecting drug users are needed - soon.
Estimated Time to Protection and Duration of Protection With Daily TDF/FTC PrEP - (05/21/14)
1. New Formulations.
1. PKPD of DRV 800mg once daily coadministered with cobicistat (abstract P_49). A fixed dose combination (FDC) tablet of darunavir (800mg) and cobicistat (150mg) has been found bioequivalent to the individual drugs. A 48 wk study of the FDC with 2 NRTIs in 313 treatment naïve or experienced persons showed a virologic response (HIV RNA < 50cpm) of 82%. Though DRV trough concentrations when DRV is given with cobicistat vs RTV are somewhat lower, the efficacy analysis showed no drop off of virologic response in the lowest quartile of trough concentrations. This FDC looks promising.
2. Fixed dose formulations of LPV/r and 3TC, and LPV/r and 3TC/ZDV (abstracts P_03 and P_04). Bioequivalence data were presented on two new FDCs with LPV/r, one with 3TC and a second with 3TC/ZDV. For both FDCs, bioequivalence was demonstrated when given under fasting conditions. Though these FDCs are unlikely to have a role in developed countries, they may be useful in developing countries.
The Clinical Pharmacology of Therapy for Hepatitis.
Jennifer J. Kiser, PharmD
Over the next year, several new agents and interferon-free combinations for the treatment of HCV will receive regulatory approval. For those that can afford and access the therapies (an important topic, but beyond the scope of this meeting report), there will be several HCV treatment options. This expanding antiviral armamentarium will present some clinical pharmacology considerations for HCV treaters. These include determining the optimal combinations of agents, dosing in special patient populations (e.g., those with advanced liver disease), the role of ribavirin, and of course, drug interactions. While dozens of compounds are in various stages of development for the treatment of HCV, only the approved or investigational agents listed in Table 1 have clinical pharmacology data included in this report.
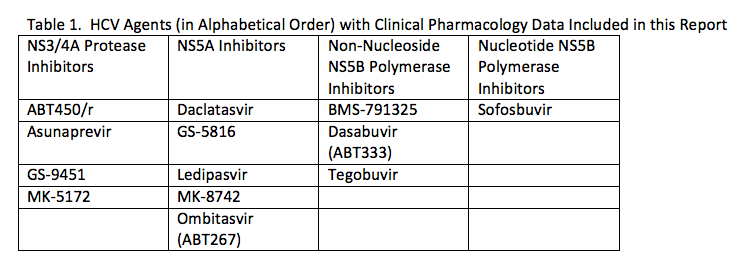
I. Drug Interactions
1. Transporter-Mediated Interactions
A focus of this year's meeting was on transporter-mediated drug interactions with current and investigational HCV agents. Analytical techniques were not available to study membrane transporters until the 1990s. Thus, transporter-mediated interactions have received far less attention than CYP enzyme-mediated interactions. However, several clinically significant drug interactions occur at the level of membrane transporters (see presentation by K Giacomini). The FDA recommends that companies test (at a minimum) whether their compounds are substrates for and inhibitors of the uptake transporters OATP1B1/1B3 (if hepatically metabolized) and OAT1 or OAT3 or OCT2 (if renally cleared) and the efflux transporters P-gp and BCRP. (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf) The data in tables 2 and 3 are a summary of transporter-mediated interactions presented at the workshop by E Mogalian (O_07), W Yeh (PP_27), T Eley, R Menon and B Kirby.
Table 2 shows the effects of investigational HCV agents on transporter substrates. Most agents are weak P-gp inhibitors and weak to moderate inhibitors of OATP1B1. There are mixed effects of these agents on BCRP. It is likely that rosuvastatin and other BCRP substrates will be contraindicated with GS-9451/ledipasvir/tegobuvir and used at a very low dose with ritonavir-boosted ABT450, ombitasvir, and dasabuvir (AbbVie 3D).
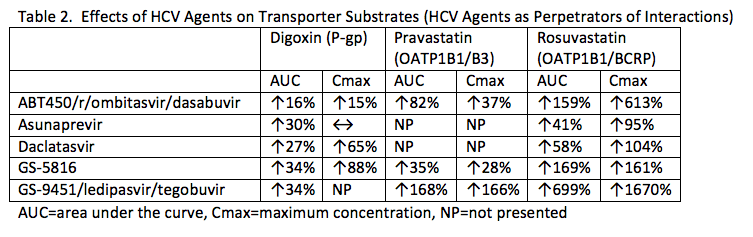
Table 3 shows the effects of other medications on the concentrations of HCV agents. Asunaprevir is clearly highly dependent on OATP1B1 for transport into the liver. Potent OATP1B1 inhibitors (which include several HIV protease inhibitors) will likely be contraindicated with this agent. GS-5816 is a CYP3A, P-gp and OATP1B1 substrate. Multi-dose rifampin reduces GS-5816 AUC and Cmax by 82% and 71%, respectively. Atazanavir increases the AUC and trough of ritonavir-boosted ABT450 by 94% and 226%, respectively.
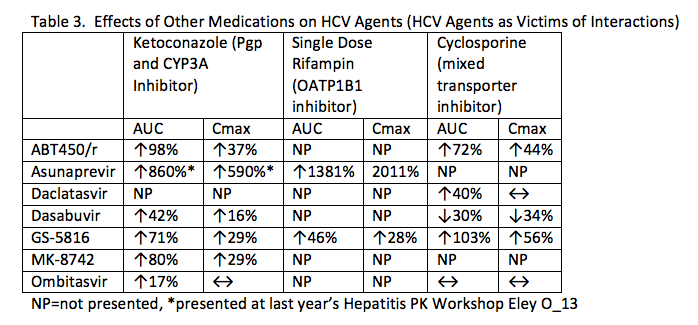
2. BMS791325 reduces the concentrations of CYP3A substrates.
Midazolam AUC was reduced by half when administered with the investigational non-nucleoside polymerase inhibitor, BMS791325, indicating this drug is a CYP3A inducer (P_22). BMS791325 has been studied in combination with asunaprevir and daclatasvir (Everson GT, et al. Gastroenterology 2014;146:420). Asunaprevir is also a CYP3A inducer (Eley AASLD 2011), so the doses of CYP3A substrates may need to be increased (even doubled) with this HCV regimen.
3. Ledipasvir and Sofosbuvir Interactions with ARV
Rates of sustained virologic response (SVR) from Hepatitis C treatment are similar in persons with HIV and HCV coinfection to persons without HIV (see presentation by M Sulkowski). Thus, the single most important consideration in the treatment of persons with HIV/HCV coinfection is the potential for antiviral drug interactions. Interaction data were presented at this year's workshop for ledipasvir (LDV) with RAL and LDV/SOF with TDF/FTC/RPV and TDF/FTC/EFV.(O_06) No interaction was observed with RPV. RAL had no effect on LDV concentrations, but LDV reduced RAL by 15%. A prior study found a 27% reduction in RAL AUC with SOF.(Kirby AASLD 2012) Thus, the combination of LDV plus SOF could conceivably reduce RAL exposures an average of 40%. TDF/FTC/EFV reduced LDV by 34%. TFV exposures when administered as TDF/FTC/EFV are increased 98% with LDV/SOF and 40% when administered as TDF/FTC/RPV. The mechanism and clinical relevance of this interaction are unclear. An increase in TFV does not occur with daclatasvir (Bifano 19th CROI 2012) or ombitasvir (see presentation by R Menon). Ongoing trials of LDV/SOF in persons with HIV/HCV coinfection include TDF, EFV, and RAL without dose adjustment. Drug interaction studies with HIV protease inhibitors, elvitegravir/cobicistat and dolutegravir are underway.
4. No effect of H2 blocker or proton pump inhibitor on ledipasvir/sofosbuvir.
Previous phase I studies found lower LDV concentrations with food or following coadministration with a proton pump inhibitor (PPI). Thus, two Phase I studies were performed to determine the effects of food and gastric acid modifiers on LDV/SOF pharmacokinetics.(P_15) LDV concentrations were similar when given fasted vs. with a moderate (600kcal) or high (1000kcal) fat meal. SOF AUC was increased 78 and 95% with a high fat and moderate fat meal, respectively vs. fasted. GS-331007 (-007) AUC was unchanged and Cmax was reduced 30% and 21% with a high and moderate fat meal, respectively relative to fasted. There was no effect of 40mg of famotidine on SOF or -007 PK. LDV AUC was unchanged by famotidine and Cmax reduced 20% when given simultaneously vs. 17% when doses were separated by 12 hours. A single 20mg dose of omeprazole had no effect on SOF, -007, or LDV PK.
15th Intl Wrkshp Clinical Pharm HIV Therapy A novel microdose approach to assess bioavailability, intestinal absorption, gut metabolism, and hepatic clearance of simeprevir in healthy volunteers - (06/16/14)
15th Intl Wrkshp Clinical Pharm HIV Therapy PBPK modeling to characterize the interplay between metabolism and transport in the disposition of simeprevir in healthy volunteers and HCV infected patients - (06/16/14)
Bristol-Myers Squibb HCV DAAs: Review of Interactions Involving Transporters - (05/28/14)
ABT-450/Ritonavir +Ombitasvir + Dasabuvir: Drug Interactions Mediated by Transporters - (05/21/14)
Gilead - Transporters: Role in Clinical Development of HCV Compounds - (05/23/14)
Drug Interactions Between Direct-Acting anti-HCV Antivirals Sofosbuvir and Ledipasvir and HIV Antiretrovirals - (05/27/14)
The Effect of Renal Impairment on Single-Dose Pharmacokinetics of Daclatasvir, an HCV NS5A Inhibitor - (05/22/14)
DDIs (drug-drug interactions) in the evolving HCV treatment landscape - (05/28/14)
Translational Studies to Understand the Mechanism of Liver Delivery by Sofosbuvir - (05/23/14)
Pharmacokinetics and Safety of Hepatitis C Virus Nonstructural Protein 5A Inhibitor MK-8742 in Cirrhotic Patients With Mild and Moderate Hepatic Insufficiency - (05/27/14)
Pharmacokinetics of Hepatitis C Virus Protease Inhibitor MK-5172 in Volunteers With Mild and Moderate Hepatic Impairment - (05/27/14)
Age and Gender Effects on the Pharmacokinetics of HCV NS5A Inhibitor MK-8742 - (05/27/14)
Pharmacokinetic Interaction of HCV NA5A Inhibitor MK-8742 and Ketoconazole in Healthy Subjects - (05/27/14)
Effect of Food and Acid Reducing Agents on the Relative Bioavailability and Pharmacokinetics of Ledipasvir/Sofosbuvir Fixed-Dose Combination Tablet - (05/27/14)
Population Pharmacokinetic Analysis of Ledipasvir (GS-5885) in Healthy and Hepatitis C Virus-Infected Subjects - (05/27/14)
Evaluation of the Effect of Ledipasvir on the QT/QTc Interval in Healthy Subjects - (05/27/14)
Evaluation of Transporter and Cytochrome P450-Mediated Drug-Drug Interactions Between Pan-Genotypic HCV NS5A Inhibitor GS-5816 and Phenotypic Probe Drugs - (05/23/14)
The Effect of Steady-state BMS-791325, a Non-nucleoside HCV NS5B Polymerase Inhibitor, on the Pharmacokinetics of Midazolam in Healthy Japanese and Caucasian Males - (05/23/14)
Assessment of Correlation of Asunaprevir With Polymorphisms in Liver Uptake Transporters (OATP1B1 and 2B1): Results of an Integrated Population PK Analyses - (05/23/14)
Asunaprevir Does Not Have an Effect on QTcF Interval in Healthy Subjects - (05/23/14)
No Clinically Meaningful Effect of Single- and Multiple-Dose Administration of Peginterferon Lambda-1a on the QTc Interval - (05/23/14)
PHARMACOKINETICS OF SIMEPREVIR, JNJ-56914845 AND RITONAVIR-BOOSTED TMC647055 WHEN CO-ADMINISTERED IN HEALTHY VOLUNTEERS - (05/22/14)
Pharmacokinetics and Safety of IDX21437, a Novel HCV Nucleotide Prodrug, in Healthy Volunteers and HCV-Infected Subjects: Results of a First-In-Human Study - (05/21/14)
II. Role of Ribavirin
RBV remains a component of several HCV treatment regimens. There is a need to define the concentration-effect associations for the drug to determine optimal dosing with direct acting agents. A retrospective analysis of RBV plasma pharmacokinetic-dynamic associations in 904 patients from the ADVANCE, ILLUMINATE, and OPTIMIZE trials found week 8 plasma concentrations to be predictive of both SVR and severe anemia (Hgb<8.5g/dL).(O_04) ROC analysis identified a week 8 plasma concentration range of 2.2-3.5 mg/L would best balance the likelihood of SVR and the risk of severe anemia. Investigators confirmed previous findings that RBV exposures were higher in the presence of telaprevir. Concentration-anemia associations were also investigated for RBV in a study of 64 HIV/HCV coinfected subjects receiving peginterferon, RBV and boceprevir.(O_05) In this study, an ROC analysis determined a RBV concentration of 2 mg/L (AUC=0.524) at week 4 was associated with a hemoglobin decline of more than 2 g/dL with a sensitivity of 41% and a specificity of 83%. These concentration targets will need to be confirmed in the context of interferon-free therapies.
III. Special Populations
1. Reduced Activity or Expression of Liver Uptake Transporter May Explain Higher Plasma Levels of HCV Drugs in Asians.
Several studies have found higher plasma exposures of HCV agents in Asian populations. This is sometimes attributed to weight differences, but Sivi Ouwerkerk-Mahadaven presented data from physiologic based pharmacokinetic modeling with simeprevir which suggests Japanese patients have a 15% lower liver volume, lower CYP3A4 abundance, and intrinsically lower OATP1B1 activity and in fact may have lower liver levels of simeprevir, but higher plasma levels. An analysis of ~1200 Asian and non-Asian subjects receiving asunaprevir (P_32) failed to identify differences in the genes encoding OATP1B1 or OATP2B1 as the cause for higher plasma levels of asunaprevir in Asians and instead postulate that there may be population differences in cell surface expression.
2. Hepatic and Renal Impairment
MK-5172 exposures are increased 62% in those with mild (Child-Pugh A) and 388% in those with moderate (Child Pugh B) hepatic impairment relative to those with no hepatic impairment.(P_37) MK5172 is in Phase 3 clinical development in combination with the NS5A inhibitor, MK-8742. Total concentrations of MK-8742 are 24% and 14% lower in patients with mild and moderate hepatic insufficiency, respectively.(P_41) This seems counterintuitive, but the same phenomenon is observed with daclatasvir (Bifano M AASLD 2011) and is likely a reflection of altered protein binding. Ideally, hepatic impairment studies should investigate pathophysiologic explanations for the altered PK by measuring free drug and/or metabolite concentrations.
Daclatasvir AUC in those with end stage renal disease (estimated glomerular filtration rate [eGFR] less than 15 mL/min/1.73m2 receiving hemodialysis), moderate renal impairment (eGFR 30-59 mL/min/1.73m2), and severe renal impairment (eGFR 15-29 mL/min/1.73m2) were 1.27, 2.10, and 1.94-fold higher, respectively compared to those with normal renal function.(P_43) The mechanism for the increased exposures is unclear given that less than 10% of a daclatasvir dose is renally eliminated. Fortunately exposures of daclatasvir are not increased by hepatic impairment (Bifano AASLD 2011), so a dual increase in exposures would not be expected in those with both advanced liver disease and renal impairment.
IV. Antiviral Concentrations in Hepatocytes
A critical question in designing HCV therapy in persons with advanced liver disease is whether blood concentrations for the HCV agents reflect their liver concentrations. Patients with advanced liver disease may have portal-systemic shunting or alterations in membrane transporter and/or phosphorylation enzyme expression or function that could affect drug concentrations in the liver. There was a case report of measuring antiviral drugs in liver tissue and hepatocytes from a 50 year old man who underwent orthotopic cadaveric liver transplantation.(P_08) Simulating a biopsy on the explanted liver, hepatocytes were isolated from cores and the active intracellular phosphorylated forms of TFV-diphosphate, FTC-triphosphate, and 3TC-triphosphate were measured. This approach is unique since fresh hepatocytes were isolated and concentration results were normalized to the hepatocyte count. This methodology may also be useful in measuring HCV agents in patients undergoing HCV treatment.
In closing, this Workshop allows individuals from industry, academia, and regulatory agencies to discuss topics and present research related to the clinical pharmacology of HIV and hepatitis therapy. A comprehensive understanding of the clinical pharmacology of antiviral drugs has revolutionized the treatment of HIV, and will likely do the same for the treatment of viral hepatitis.
|
| |
|
|
|
|
|