 |
|
|
| |
Sofosbuvir With Velpatasvir (GS-5816) in Treatment-Naive Noncirrhotic Patients With Genotype 1 to 6 Hepatitis C Virus Infection: A Randomized Trial
|
| |
| |
Download the PDF here
"This is, to our knowledge, the first demonstration of a DAA combination with pangenotypic activity against HCV genotypes 1 to 6."
Ann Intern Med. Published online 10 November 2015
Gregory T. Everson, MD; William J. Towner, MD; Mitchell N. Davis, DO; David L. Wyles, MD; Ronald G. Nahass, MD; Paul J. Thuluvath, MD; Kyle Etzkorn, MD; Federico Hinestrosa, MD; Myron Tong, MD, PhD; Mordechai Rabinovitz, MD; John McNally, PhD; Diana M. Brainard, MD; Lingling Han, PhD; Brian Doehle, PhD; John G. McHutchison, MD; Timothy Morgan, MD; Raymond T. Chung, MD; and Tram T. Tran, MD
Abstract
Background: Effective, pangenotypic treatments for hepatitis C virus (HCV) infection are needed.
Objective: To assess the safety and efficacy of sofosbuvir with velpatasvir in patients infected with HCV genotypes 1 to 6.
Design: Randomized, phase 2, open-label study. (ClinicalTrials.gov: NCT01858766)
Setting: 48 U.S. sites.
Patients: 377 treatment-naive noncirrhotic patients. In part A, patients infected with HCV genotypes 1 to 6 were randomly assigned to sofosbuvir, 400 mg, with velpatasvir, 25 or 100 mg, for 12 weeks. In part B, patients with genotype 1 or 2 HCV infection were randomly assigned to sofosbuvir, 400 mg, and velpatasvir, 25 or 100 mg, with or without ribavirin for 8 weeks.
Measurements: Sustained virologic response at 12 weeks (SVR12).
Results: In part A, SVR12 rates were 96% (26 of 27) with velpatasvir, 25 mg, and 100% (28 of 28) with velpatasvir, 100 mg, for genotype 1; 93% (25 of 27) in both groups for genotype 3; and 96% (22 of 23) with velpatasvir, 25 mg, and 95% (21 of 22) with velpatasvir, 100 mg, for genotypes 2, 4, 5, and 6. In part B, for genotype 1, SVR12 rates were 87% (26 of 30) with velpatasvir, 25 mg; 83% (25 of 30) with velpatasvir, 25 mg, plus ribavirin; 90% (26 of 29) with velpatasvir, 100 mg; and 81% (25 of 31) with velpatasvir, 100 mg, plus ribavirin. For genotype 2, SVR12 rates were 77% (20 of 26) with velpatasvir, 25 mg; 88% (22 of 25) with velpatasvir, 25 mg, plus ribavirin; 88% (23 of 26) with velpatasvir, 100 mg; and 88% (23 of 26) with velpatasvir, 100 mg, plus ribavirin. Adverse events included fatigue (21%), headache (20%), and nausea (12%). One patient committed suicide.
Limitation: The study was Open-label, no inferential statistics were planned, and sample sizes were small.
Conclusion: Twelve weeks of sofosbuvir, 400 mg, and velpatasvir, 100 mg, was well-tolerated and resulted in high SVR in patients infected with HCV genotypes 1 to 6.
Primary Funding Source: Gilead Sciences.
Up to 150 million persons worldwide are chronically infected with hepatitis C virus (HCV) (1). Chronic HCV infection causes slowly progressive liver fibrosis, which can lead to cirrhosis, hepatic decompensation, and hepatocellular carcinoma (2, 3). Chronic HCV infection is curable, as measured by the end point of sustained virologic response (SVR), which is defined as undetectable HCV RNA 12 or 24 weeks after the end of treatment. For many years, however, HCV treatment regimens were based on recombinant interferon-α, a poorly tolerated drug with moderate efficacy (4). The recent advent of drugs that directly target HCV viral replication has revolutionized treatment of HCV infection. The first such direct-acting antiviral agents (DAAs), the protease inhibitors telaprevir and boceprevir, were approved in combination with peginterferon and ribavirin in 2009, but these regimens are no longer recommended (5). With a second generation of DAAs, effective interferon-free treatment of HCV infection is now available. However, existing DAA combination regimens are not uniformly effective across the 6 HCV genotypes. The most effective regimens are for genotype 1 HCV, which accounts for approximately 46% of infections worldwide (6). Genotypes 2 and 3 HCV, the next most common genotypes, were historically grouped together in treatment guidelines, but recent studies have shown that infection with genotype 3, especially in patients with cirrhosis, is more difficult to treat (7, 8). Many interferon-free combinations are now available for these and the other 3 HCV genotypes (9, 10), but clinicians must take into account the genotype and even the subtype as well as patterns of antiviral resistance in the choice of a regimen. The development of a single, short-duration regimen that is safe and effective in all HCV genotypes would greatly simplify treatment and would reduce the need for pretreatment testing and on-treatment monitoring.
Sofosbuvir is a pangenotypic HCV NS5B polymerase inhibitor approved in the United States and other regions for the treatment of HCV infection (11). Velpatasvir is a novel inhibitor of the HCV NS5A protein that has demonstrated pangenotypic antiviral activity in vitro (12) and in a 3-day monotherapy study in patients infected with genotypes 1, 2, 3, and 4 HCV (13). Drug interaction studies showed no clinically significant interactions between sofosbuvir and velpatasvir (14), and both can be administered once daily (15).
The clinical potential of the combination of sofosbuvir and velpatasvir is suggested by their nonoverlapping viral targets, the proven success of sofosbuvir in combination with other NS5A inhibitors, and the in vitro potency of velpatasvir. We assessed the safety and efficacy of 8 or 12 weeks of velpatasvir coadministered with sofosbuvir. In addition, we examined the effect of adding ribavirin for patients receiving 8 weeks of treatment.
Methods
Study Overview
Enrollment for this phase 2, multicenter, randomized, open-label study began on 22 August 2013, and data collection for the primary efficacy end point was completed on 12 August 2014 (Appendix Table). As originally designed, the study consisted of 6 groups of treatment-naive patients receiving 12 weeks of treatment: approximately 50 patients with genotype 1 HCV infection (groups 1 and 2); 50 with genotype 3 infection (groups 3 and 4); and 50 with genotype 2, 4, 5, or 6 infection (groups 5 and 6). Because interim analyses of results from these 6 groups (part A) indicated that patients with genotype 1 and 2 HCV infection achieved high rates of SVR at 12 weeks (SVR12), we amended the protocol to include part B, in which we evaluated 8 weeks of treatment with and without ribavirin in patients with genotype 1 infection (groups 7 to 10) and genotype 2 infection (groups 11 to 14).
Setting and Participants
The study was conducted at 48 sites in the United States. Patients were recruited partly through a posting of study details on ClinicalTrials.gov and partly through referral by their treating physicians.
Treatment-naive noncirrhotic adults (aged ≥18 years) with HCV RNA levels greater than 10 000 IU/mL were eligible. The HCV RNA genotype was determined by using the Siemens VERSANT HCV Genotype Assay, version 2 (LiPA 2.0), or, if results were inconclusive, the TRUGENE HCV 5'NC Genotyping Kit was used with the OpenGene DNA Sequencing System. Absence of cirrhosis was established by liver biopsy within 2 years of screening; a FibroTest score of 0.48 or less and an aspartate aminotransferase-platelet ratio index of 1 or less during screening; or a Fibroscan score of 12.5 kPa or less within 6 months of baseline. Exclusion criteria were previous treatment for HCV infection, hepatic decompensation or co-infection with hepatitis B virus or HIV, aminotransferase levels greater than 10 times the upper limit of normal (ULN), direct bilirubin level greater than 1.5 times the ULN, platelet count less than 90 x 109 cells/L, hemoglobin A1c level greater than 8.5%, creatinine clearance less than 60 mL/min (as calculated by the Cockcroft-Gault equation), hemoglobin level less than 110 g/L for female patients or less than 120 g/L for male patients, albumin level less than 30 g/L, and prothrombin time (international normalized ratio) greater than 1.5 times the ULN.
All patients provided written informed consent before screening. The protocol was approved by institutional ethics committees at all study sites. The study was conducted in accordance with good clinical practice and the Declaration of Helsinki and was registered with ClinicalTrials.gov (NCT01858766).
Randomization and Interventions
In part A, patients in each of the 3 HCV genotype categories were randomly assigned in a 1:1 ratio to receive 25 or 100 mg of velpatasvir once daily for 12 weeks. All patients received 400 mg of sofosbuvir once daily. Patients with genotype 1 HCV infection were stratified by HCV subtype (1a vs. 1b). Patients in groups 5 and 6 were stratified by HCV genotype.
In part B, patients in each of the 2 HCV genotype categories were randomly assigned in a 1:1:1:1 ratio to receive 25 or 100 mg of velpatasvir once daily for 8 weeks or 25 or 100 mg of velpatasvir once daily plus ribavirin in a divided daily dose. All patients received 400 mg of sofosbuvir once daily for 8 weeks. Patients with genotype 1 HCV infection were stratified by HCV subtype (1a vs. 1b).
Sofosbuvir was administered to all patients once daily in a 400-mg tablet. Velpatasvir was administered once daily in 25- or 100-mg tablets. Ribavirin was administered twice daily at a dose of 1000 mg/d (three 200-mg tablets in the morning and two 200-mg tablets in the evening) in patients with a total body weight less than 75 kg and 1200 mg/d (three 200-mg tablets twice daily) in those with a total body weight of 75 kg or greater (16).
Patient randomization was managed by using an interactive Web response system (Bracket). A statistician employed by the sponsor (L.H.) generated the randomization code using a SAS program, which was validated by another statistician employed by the sponsor. Randomization was stratified by HCV genotype and used a block size of 4. Investigators, patients, and trial personnel were not blinded to treatment assignments at any point.
Outcomes and Follow-up
The primary efficacy outcome measure was SVR12, defined as a serum HCV RNA level below the lower limit of quantification (LLOQ) 12 weeks after completion of treatment. The HCV RNA concentration was measured using the Roche COBAS TaqMan HCV Test (v2.0) with the High Pure System, with an LLOQ of 25 IU/mL.
On-treatment virologic failure was defined as an HCV RNA level of at least 25 IU/mL after 8 weeks of therapy. Relapse was defined as an HCV RNA level of at least the LLOQ during the posttreatment follow-up in patients who achieved a level below the LLOQ by the end of treatment. Levels of HCV RNA were measured at posttreatment weeks 4, 8, 12, and 24.
Deep sequencing of the HCV NS5A and NS5B genes was performed from pretreatment plasma samples from all enrolled patients and from posttreatment samples from all patients with virologic failure. NS5A resistance-associated variants (RAVs) were defined as changes from a genotype-specific reference sequence at amino acid positions 28, 30, 31, 32, 58, 92, and 93. NS5B RAVs were defined as any change from the HCV reference sequence at positions 96, 142, 159, 282, 289, 320, and 321.
Statistical Analysis
The analysis set for efficacy and safety included randomly assigned patients who received at least 1 dose of the study drug. No inferential statistics or statistical comparisons were planned or conducted for efficacy or safety data. A 2-sided 95% CI for the proportion of patients with SVR12 within treatment groups was calculated by using the Clopper-Pearson method, which provides the exact CI based on the binomial distribution rather than an approximation to the binomial distribution (17). Patients with missing HCV RNA values for any reason at posttreatment week 12 and after were counted as treatment failures. No formal power or sample size calculations were used to determine group size. With a sample size of 25 patients in each treatment group, a 2-sided 95% exact CI was calculated to extend, at most, 41% in length. We performed all statistical analyses with SAS, version 9.2 (SAS Institute).
Role of the Funding Source
Gilead Sciences funded this study and was involved in the design of the study; collection, analysis, and interpretation of the data; and the writing of the report.
Results
Baseline Characteristics
A total of 469 patients were screened, of whom 377 were randomly assigned and received treatment at 48 sites in the United States, with a median of 8 patients enrolled per site (range, 1 to 26 patients). All 377 patients were included in the final analysis (Figure). Table 1 shows demographic, disease, and baseline characteristics in the treatment groups.
Virologic Response
On-Treatment Response
All patients had HCV RNA levels below the LLOQ after 4 weeks of treatment, except for 1 infected with genotype 3 HCV who was randomly assigned to receive sofosbuvir with velpatasvir, 25 mg, for 12 weeks (group 3). This patient had a reduction in HCV RNA level of 5 log10 IU/mL after 4 weeks of treatment but had an HCV RNA level above the LLOQ after 8 weeks, and treatment was discontinued in this patient for virologic nonresponse per protocol (Figure).
Sustained Virologic Response
Overall, among the 377 patients randomly assigned and treated, 337 (89%) had SVR12. Of these, 324 returned for the posttreatment week 24 visit, at which 323 (>99%) had SVR (the remaining patient is described in the next paragraph). Of the 13 remaining patients, 6 were lost to follow-up after posttreatment week 12 and 7 attended visits after posttreatment week 12 but outside the window for posttreatment week 24; all 7 had HCV RNA levels below the LLOQ at these visits.
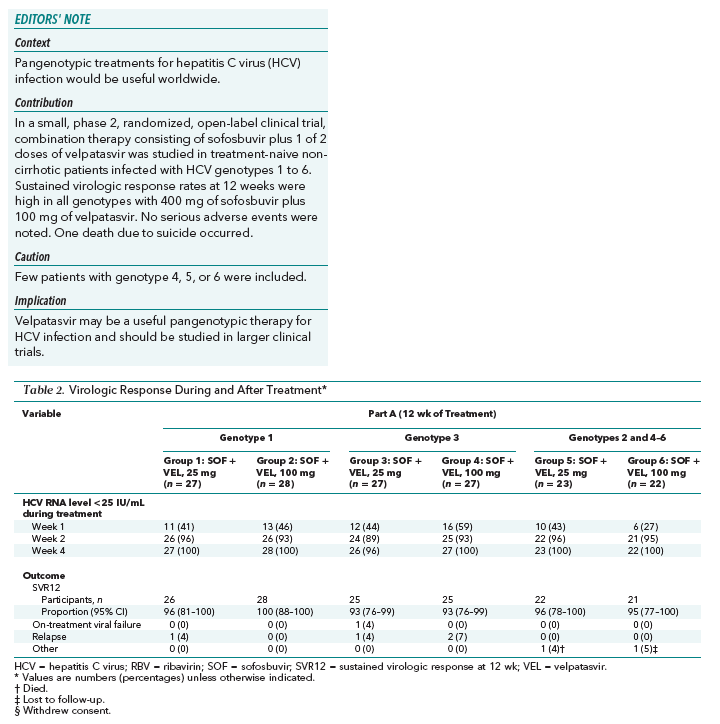
In part A, among patients with genotype 1 HCV infection, the SVR12 rate was 96% (26 of 27) in those receiving sofosbuvir with velpatasvir, 25 mg, for 12 weeks (group 1) and 100% (28 of 28) in those receiving sofosbuvir with velpatasvir, 100 mg, for 12 weeks (group 2) (Table 2). One patient with genotype 1 HCV infection who received sofosbuvir with velpatasvir, 25 mg, for 12 weeks had virologic relapse 4 weeks after the end of treatment. One patient, a 29-year-old white woman with genotype 1a HCV infection and a baseline HCV RNA level of 6.8 log10 IU/mL who was randomly assigned to receive sofosbuvir with velpatasvir, 25 mg, for 12 weeks (group 1), achieved SVR12 but was viremic 24 weeks after completion of treatment. Comparison of pretreatment and posttreatment HCV sequences indicated that a new HCV infection had occurred rather than a reemergence of the pretreatment viral population.
Among patients with genotype 3 HCV infection, the SVR12 rate was 93% (25 of 27) in those receiving sofosbuvir with velpatasvir, 25 mg, for 12 weeks (group 3) and 93% (25 of 27) in those receiving sofosbuvir with velpatasvir, 100 mg, for 12 weeks (group 4). Two patients with genotype 3 HCV infection who were administered sofosbuvir with velpatasvir, 25 mg, for 12 weeks (group 3) did not achieve SVR12; 1 had virologic nonresponse, and 1 had virologic relapse after treatment. Two patients with genotype 3 HCV infection who received sofosbuvir with velpatasvir, 100 mg, for 12 weeks (group 4) did not achieve SVR12; both had posttreatment virologic relapse. An HCV sequence analysis revealed that a new HCV infection (genotype 2b) had occurred in 1 of these patients.
The SVR12 rate in patients with genotype 2, 4, 5, or 6 HCV infection receiving sofosbuvir with velpatasvir, 25 mg, for 12 weeks (group 5) was 96% (22 of 23); 1 patient with genotype 2 HCV infection committed suicide before posttreatment week 12. The SVR12 rate in patients with genotype 2, 4, 5, or 6 HCV infection receiving sofosbuvir with velpatasvir, 100 mg, for 12 weeks (group 6) was 95% (21 of 22); 1 patient with genotype 4 HCV infection completed treatment and was subsequently lost to follow-up.
In part B, SVR12 rates among patients with genotype 1 HCV infection were 87% (26 of 30) for those receiving velpatasvir, 25 mg; 83% (25 of 30) for those receiving velpatasvir, 25 mg, plus ribavirin; 90% (26 of 29) for those receiving velpatasvir, 100 mg; and 81% (25 of 31) for those receiving velpatasvir, 100 mg, plus ribavirin. All virologic failures were due to posttreatment relapse. Two patients did not complete posttreatment assessments; one was a patient treated with sofosbuvir with velpatasvir, 100 mg, plus ribavirin (group 10), and the other was a patient treated with sofosbuvir with velpatasvir, 25 mg (group 7), who discontinued treatment after 8 days because of adverse events and then withdrew consent from the study.
Rates of SVR12 among patients with genotype 2 HCV infection were 77% (20 of 26) with velpatasvir, 25 mg; 88% (22 of 25) with velpatasvir, 25 mg, plus ribavirin; 88% (23 of 26) with velpatasvir, 100 mg; and 88% (23 of 26) with velpatasvir, 100 mg, plus ribavirin. All virologic failures were due to posttreatment relapse. One patient with genotype 2 HCV infection treated with velpatasvir, 25 mg, plus ribavirin for 8 weeks did not complete posttreatment assessments.
Viral Resistance Assessment
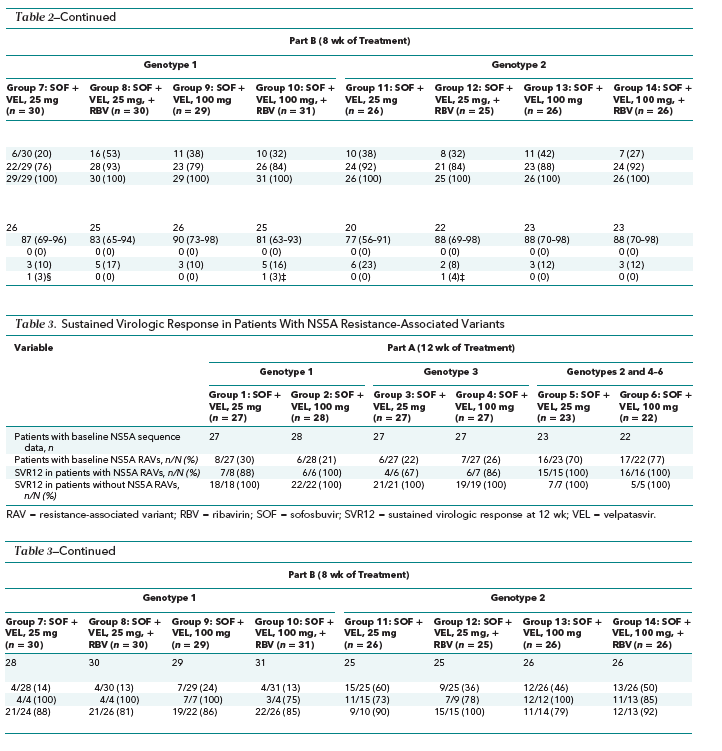
Of the 377 patients enrolled, 375 and 372 had sequencing data for HCV NS5A and NS5B, respectively. The prevalence of pretreatment NS5A RAVs detected with a 15% cutoff was 34% (128 of 375) overall and 18% (25 of 142), 23% (7 of 31), 48% (58 of 122), and 24% (13 of 54) in patients with genotype 1a, 1b, 2, and 3 HCV infection, respectively. Overall rates of SVR were 90% in patients with pretreatment RAVs and 92% in those without. In part A, SVR12 rates were 100% in patients without pretreatment RAVs, regardless of genotype (Table 3). Among the 6 patients treated for 12 weeks who did not achieve SVR, 1 had virologic nonresponse, 3 relapsed, and 2 had HCV reinfection. All 4 patients with virologic failure (excluding the 2 with reinfection) had pretreatment and posttreatment NS5A RAVs, and 1 had the NS5B RAV N142T detected before and after treatment.
In part B, we found no consistent pattern of rates of SVR after 8 weeks of treatment with or without pretreatment RAVs (Table 3). In the patients infected with HCV genotype 1 in groups 7 to 10, rates of SVR12 were higher in those with pretreatment RAVs. In contrast, in the patients with HCV genotype 2 infection and pretreatment RAVs, rates of SVR12 were lower in groups 11, 12, and 14 but higher in group 13. The numbers of patients with baseline RAVs within each treatment group are too small to draw firm conclusions about the role of baseline RAVs in defining treatment response.
Seventeen of 372 patients (5%) had pretreatment NS5B RAVs, of whom 15 (88%) achieved SVR12. The NS5B RAV S282T was not observed in any patient in this study.
Safety Results
Table 4 shows adverse events and laboratory abnormalities in the study. Overall, 262 (69%) patients reported at least 1 adverse event. Adverse events experienced by more than 10% of patients were fatigue (21%), headache (20%), and nausea (12%). We found no difference in the type or incidence of adverse events between treatment regimens with respect to velpatasvir dose or treatment duration.
Incidence of fatigue, insomnia, and rash was higher in patients treated with ribavirin-containing regimens. One patient, a 19-year-old white woman with genotype 1 HCV infection who was receiving 8 weeks of sofosbuvir plus velpatasvir, 25 mg (group 1), had mild abdominal pain, mild palpitations, and moderate dizziness on treatment day 6. The investigator assessed the events as related to a study drug, and treatment was discontinued the following day. All of these events resolved by day 2 of follow-up. Eight serious adverse events were reported in 7 (2%) patients, and none were assessed by the reporting investigator as related to the study drug. One death occurred in the study: A 36-year-old man with genotype 2 HCV infection and preexisting psychiatric disease committed suicide after completing 12 weeks of treatment with sofosbuvir and velpatasvir, 25 mg (group 5).
Incidence of decreased hemoglobin levels and elevated bilirubin levels was low (n = 4 [1%] and 2 [0.5%], respectively) and occurred in the patients receiving ribavirin. One patient had a reduction in neutrophil count to a nadir of 0.570 x 109 cells/L, from a pretreatment count of 0.790 x 109 cells/L. We found no abnormalities in lymphocyte, platelet, or leukocyte counts.
Discussion
In this phase 2 study of treatment-naive noncirrhotic patients, high rates of SVR12 were achieved across HCV genotypes 1 to 6 with 12 weeks of sofosbuvir and velpatasvir. Virologic failure was rare among patients treated for 12 weeks, occurring in only 1 of 55 patients with HCV genotype 1 infection, 3 of 54 with genotype 3 infection, and 0 of 45 with genotype 2, 4, 5, or 6 infection. The nonresponse of 1 patient with HCV genotype 3 infection and the relapse in a patient with HCV genotype 1 infection, both of whom were receiving 25 mg of velpatasvir, suggest that the 100-mg dose could have a clinical advantage over the 25-mg dose when administered in combination with sofosbuvir for 12 weeks.
Rates of SVR were lower with 8 weeks versus 12 weeks of treatment in patients with HCV genotype 1 or 2 infection. The small sample size and limited number of virologic failures in each treatment group precluded analysis of the association between viral load and virologic failure. The high SVR rates achieved with 12 weeks of treatment, even in patients with pretreatment RAVs, may indicate that the 12-week regimen is more efficacious for patients with negative predictors, such as baseline resistance.
Overall, treatment with sofosbuvir and velpatasvir with or without ribavirin was well-tolerated, with low rates of serious adverse events and only 1 discontinuation due to adverse events. Patients administered ribavirin-containing regimens had higher incidence of ribavirin-associated toxicities, such as fatigue, insomnia, and rash, and laboratory abnormalities associated with ribavirin-induced hemolysis, such as decreased hemoglobin levels and elevated bilirubin levels (16).
This is, to our knowledge, the first demonstration of a DAA combination with pangenotypic activity against HCV genotypes 1 to 6. The SVR rates in this study for treatment-naive noncirrhotic patients were similar to those observed with standard-of-care regimens. In the ION-1 phase 3 trial, noncirrhotic patients with genotype 1 HCV infection who were treated with ledipasvir plus sofosbuvir for 12 weeks had an SVR12 rate of 99%, compared with the 100% rate we observed with 12 weeks of sofosbuvir and velpatasvir, 100 mg (18). Treatment-naive noncirrhotic patients with genotype 2 HCV infection who received sofosbuvir and ribavirin for 12 weeks in the VALENCE trial had an SVR rate of 97%, compared with the SVR rate of 100% we observed with 12 weeks of sofosbuvir and velpatasvir, 100 mg (19). In the VALENCE trial, treatment-naive noncirrhotic patients with genotype 3 infection who received sofosbuvir and ribavirin for 24 weeks had an SVR12 rate of 95%, which is similar to the 93% rate we observed in patients receiving sofosbuvir and velpatasvir, 100 mg, for 12 weeks (13). Reducing the duration of therapy from 24 to 12 weeks for a common and difficult-to-treat genotype, such as genotype 3, would be an important advancement.
A single regimen with pangenotypic efficacy could obviate the need for HCV genotyping before initiation of treatment. The high efficacy demonstrated with coadministration of sofosbuvir and velpatasvir, 100 mg, for 12 weeks across all HCV genotypes evaluated in this study supports the development of a single, uniform pangenotypic regimen with these DAAs. Although ribavirin was not evaluated as part of the 12-week regimens in this study, the high SVR achieved without it suggests that it would not be required for optimal efficacy, further reducing the complexity of treatment by eliminating the need for safety monitoring and pregnancy precautions necessitated by the hemolytic and teratogenic side effects of ribavirin.
Limitations of the current study are the open-label design and the exclusion of patients with cirrhosis and prior treatment failure. Rates of SVR12 have historically been lower in patients with cirrhosis compared with those without cirrhosis and prior treatment failure, even with the recently approved all-oral DAA combinations, and it is therefore important that the safety and efficacy of the combination of sofosbuvir and velpatasvir be evaluated in these populations. Other limitations are the lack of planned inferential statistics and the small number of patients with genotype 4, 5, or 6 HCV infection (the latter due to enrollment being restricted to the United States, where these genotypes are rare).
In summary, sofosbuvir with velpatasvir, 100 mg, for 12 weeks was well-tolerated and resulted in high SVR12 rates in noncirrhotic patients infected with HCV genotypes 1 to 6. These data support the coformulation of a fixed-dose combination tablet of sofosbuvir and velpatasvir, 100 mg, for evaluation in larger phase 3 studies.
|
| |
|
|
|
|
|