 |
|
|
| |
Sofosbuvir and Velpatasvir for HCV in Patients with Decompensated Cirrhosis (ASTRAL-4)
|
| |
| |
Download the PDF here
Download the PDF here
NEJM Nov 16 2015
"We conducted a phase 3, open-label trial to assess the efficacy and safety of a fixed dose of sofosbuvir-velpatasvir with or without ribavirin for 12 weeks or sofosbuvir-velpatasvir for 24 weeks in patients infected with HCV genotypes 1 through 6 and with decompensated cirrhosis....patients were randomly assigned in a 1:1:1 ratio to receive a fixed-dose combination tablet containing 400 mg of sofosbuvir and 100 mg of velpatasvir, administered orally once daily for 12 weeks; sofosbuvir-velpatasvir plus ribavirin once daily for 12 weeks; or sofosbuvir-velpatasvir once daily for 24 weeks.
Overall, 60% of patients had HCV genotype 1a, 18% genotype 1b, 4% genotype 2, 15% genotype 3, 3% genotype 4, and less than 1% genotype 6; no patients had genotype 5. A total of 6% of patients were black, and 55% had received prior treatment for HCV infection. The median baseline CPT score was 8 (range, 5 to 10), the median baseline MELD score was 10 (range, 6 to 24), and the median creatinine clearance was 84.7 ml per minute (range, 15 to 198). The majority of patients (95%) had a baseline MELD score of 15 or less. All the patients had CPT class B cirrhosis at screening, but 27 patients (10%) had CPT class A or CPT class C cirrhosis at treatment baseline, which reflects the dynamic changes in CPT scoring in this population.
In conclusion, treatment with the fixed-dose combination tablet of sofosbuvir-velpatasvir with or without ribavirin for 12 weeks and with sofosbuvir-velpatasvir for 24 weeks resulted in a high rate of sustained virologic response and early improvements in hepatic function in patients with decompensated cirrhosis caused by HCV of all genotypes. Whether the early improvements in liver function translate to long-term clinical benefits remains to be determined, and future trials are required to assess this regimen in patients with more severe degrees of liver decompensation.
A total of 9 patients discontinued study treatment prematurely because of an adverse event: 1 of 90 patients (1%) who received sofosbuvir-velpatasvir for 12 weeks, 4 of 87 patients (5%) who received sofosbuvir-velpatasvir and ribavirin, and 4 of 90 patients (4%) who received sofosbuvir-velpatasvir for 24 weeks (Table 3). No adverse event that led to discontinuation of a study drug was reported in more than 1 patient. Serious adverse events occurred in 19% of patients who received sofosbuvir-velpatasvir for 12 weeks, 16% of those who received sofosbuvir-velpatasvir plus ribavirin, and 18% of those who received sofosbuvir-velpatasvir for 24 weeks. The most common serious adverse events were hepatic encephalopathy and sepsis (with each event occurring in 5 patients across groups). Nine deaths occurred during the study. Two patients died after discontinuing study treatment but within 30 days after the end of treatment, and seven patients died more than 30 days after the end of treatment. Most of the deaths were due to complications of end-stage liver disease (i.e., liver failure, sepsis, or multiorgan failure). The nine deaths were evenly divided among the three treatment groups; none were considered to be related to therapy by the investigator. Overall, the rate of adverse events was similar among the three treatment groups. Patients who received sofosbuvir-velpatasvir plus ribavirin had a slightly higher rate of adverse events overall and a substantially higher rate of certain events known to be associated with ribavirin therapy than those who received sofosbuvir-velpatasvir alone. Close monitoring of patients with decompensated liver disease who are receiving a combination of direct-acting antiviral agents remains important in light of the limited safety information concerning these new drugs in this sicker population."
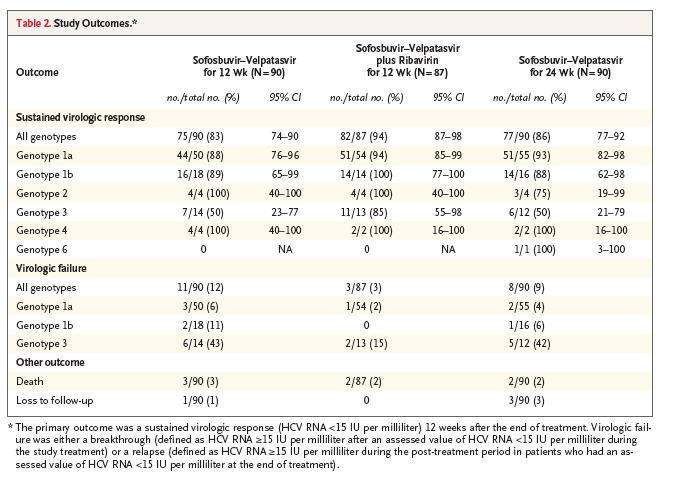
Figure 1. Changes in CPT and MELD Scores from Baseline.
Shown are the changes from baseline to post-treatment week 12 in scores on the Child-Pugh-Turcotte (CPT) scale (ranging from 5 to 15, with higher values indicating more advanced liver disease) (Panel A) and scores on the Model for End-Stage Liver Disease (MELD) scale (ranging from 6 to 40, with higher values indicating more advanced liver disease) for patients with available outcome data in all three treatment groups combined. (The study was not designed or powered to detect significant differences in rates of sustained virologic response among the three treatment groups, although a comparison of such rates was performed in a post hoc pairwise analysis.) For the MELD score, 223 patients had a baseline score of less than 15 (Panel B) and 27 had a baseline score of 15 or more (Panel C). Columns indicate the percentage of patients categorized according to the extent of change from baseline. Seventeen patients who did not undergo CPT or MELD assessments at post-treatment week 12 are omitted from the analysis. Percentages may not sum to totals in subgroups because of rounding.
Sofosbuvir and Velpatasvir for HCV in Patients with Decompensated Cirrhosis (ASTRAL-4)
Michael P. Curry, M.D., Jacqueline G. O'Leary, M.D., M.P.H., Natalie Bzowej, M.D., Andrew J. Muir, M.D., Kevin M. Korenblat, M.D., Jonathan M. Fenkel, M.D., K. Rajender Reddy, M.D., Eric Lawitz, M.D., Steven L. Flamm, M.D., Thomas Schiano, M.D., Lewis Teperman, M.D., Robert Fontana, M.D., Eugene Schiff, M.D., Michael Fried, M.D., Brian Doehle, Ph.D., Di An, Ph.D., John McNally, Ph.D., Anu Osinusi, M.D., Diana M. Brainard, M.D., John G. McHutchison, M.D., Robert S. Brown, Jr., M.D., M.P.H., and Michael Charlton, M.D. for the ASTRAL-4 Investigators
NEJM November 16, 2015
ABSTRACT
Background
As the population that is infected with the hepatitis C virus (HCV) ages, the number of patients with decompensated cirrhosis is expected to increase.
Methods
We conducted a phase 3, open-label study involving both previously treated and previously untreated patients infected with HCV genotypes 1 through 6 who had decompensated cirrhosis (classified as Child-Pugh-Turcotte class B). Patients were randomly assigned in a 1:1:1 ratio to receive the nucleotide polymerase inhibitor sofosbuvir and the NS5A inhibitor velpatasvir once daily for 12 weeks, sofosbuvir-velpatasvir plus ribavirin for 12 weeks, or sofosbuvir-velpatasvir for 24 weeks. The primary end point was a sustained virologic response at 12 weeks after the end of therapy.
Results
Of the 267 patients who received treatment, 78% had HCV genotype 1, 4% genotype 2, 15% genotype 3, 3% genotype 4, and less than 1% genotype 6; no patients had genotype 5. Overall rates of sustained virologic response were 83% (95% confidence interval [CI], 74 to 90) among patients who received 12 weeks of sofosbuvir-velpatasvir, 94% (95% CI, 87 to 98) among those who received 12 weeks of sofosbuvir-velpatasvir plus ribavirin, and 86% (95% CI, 77 to 92) among those who received 24 weeks of sofosbuvir-velpatasvir. Post hoc analysis did not detect any significant differences in rates of sustained virologic response among the three study groups. Serious adverse events occurred in 19% of patients who received 12 weeks of sofosbuvir-velpatasvir, 16% of those who received 12 weeks of sofosbuvir-velpatasvir plus ribavirin, and 18% of those who received 24 weeks of sofosbuvir-velpatasvir. The most common adverse events were fatigue (29%), nausea (23%), and headache (22%) in all patients and anemia (31%) in the patients receiving ribavirin.
Conclusions
Treatment with sofosbuvir-velpatasvir with or without ribavirin for 12 weeks and with sofosbuvir-velpatasvir for 24 weeks resulted in high rates of sustained virologic response in patients with HCV infection and decompensated cirrhosis. (Funded by Gilead Sciences; ASTRAL-4 ClinicalTrials.gov number, NCT02201901.)
The number of patients with decompensated cirrhosis caused by chronic infection with the hepatitis C virus (HCV) is projected to rise in the coming decade.1 For many years, the only treatment option for such patients was liver transplantation. Recently, however, clinical trials of newly approved direct-acting antiviral agents have shown that it is possible to treat HCV infection safely and effectively in patients with decompensated cirrhosis and that successful treatment is associated with early improvement in liver function.2-11 The possible long-term benefits of treatment on existing liver disease remain unknown. The only regimen that is currently approved for the treatment of HCV infection in patients with decompensated cirrhosis is 24 weeks of ledipasvir-sofosbuvir plus ribavirin, which is approved in Europe for patients with HCV genotypes 1 and 4.12 A highly effective regimen to treat HCV infection of all genotypes in patients with decompensated liver disease that has acceptable side effects would address a significant unmet medical need.
The NS5B nucleotide inhibitor sofosbuvir is approved for the treatment of HCV infection in combination with other agents.13,14 Velpatasvir (formerly known as GS-5816, Gilead Sciences) is an investigational inhibitor of the HCV NS5A protein with antiviral activity against all HCV genotypes.15-17 The combination of velpatasvir and sofosbuvir with or without ribavirin provided high rates of sustained virologic response in patients with all HCV genotypes in phase 2 clinical trials.18,19 In the phase 3 ASTRAL-1, ASTRAL-2, and ASTRAL-3 trials (now published in the Journal),20,21 treatment with sofosbuvir-velpatasvir in a fixed-dose combination tablet for 12 weeks resulted in high rates of sustained virologic response among patients with HCV genotypes 1 through 6 without cirrhosis or with compensated cirrhosis.
We conducted a phase 3, open-label trial to assess the efficacy and safety of a fixed dose of sofosbuvir-velpatasvir with or without ribavirin for 12 weeks or sofosbuvir-velpatasvir for 24 weeks in patients infected with HCV genotypes 1 through 6 and with decompensated cirrhosis.
Methods
Patients
We enrolled patients at 47 sites in the United States from August 19, 2014, through December 19, 2014. Eligible patients were 18 years of age or older with chronic HCV infection of any genotype and decompensated cirrhosis, classified as Child-Pugh-Turcotte (CPT) class B (a score of 7 to 9 on a scale ranging from 5 to 15, with higher values indicating more advanced liver disease). Patients who had undergone liver transplantation were not eligible. For patients awaiting liver transplantation, study treatment was required to start at least 12 weeks before the expected date of transplantation. Patients who had received previous treatment with any NS5A inhibitor or nucleotide analogue NS5B inhibitor were excluded, as were patients with a platelet count of 30,000 per cubic millimeter or less or a creatinine clearance of less than 50 ml per minute (as calculated by the Cockcroft-Gault equation). All patients provided written informed consent. Full eligibility criteria are provided in the protocol, available with the full text of this article at NEJM.org.
Study Design
In this multicenter, open-label trial, patients were randomly assigned in a 1:1:1 ratio to receive a fixed-dose combination tablet containing 400 mg of sofosbuvir and 100 mg of velpatasvir, administered orally once daily for 12 weeks; sofosbuvir-velpatasvir plus ribavirin once daily for 12 weeks; or sofosbuvir-velpatasvir once daily for 24 weeks. Ribavirin was administered orally with food twice daily, with the dose determined according to body weight (1000 mg daily in patients with a body weight of <75 kg and 1200 mg daily in patients with a body weight ≥75 kg). Randomization was stratified according to HCV genotype.
Study Assessments
Screening assessments included serum HCV RNA levels, IL28B genotyping, and standard laboratory and clinical tests, including those for the calculation of the CPT score and the Model for End-Stage Liver Disease (MELD) score (ranging from 6 to 40, with higher scores indicating more advanced liver disease). Serum HCV RNA was measured with the use of the COBAS AmpliPrep/COBAS TaqMan HCV Quantitative Test, version 2.0 (Roche Molecular Systems), with a lower limit of quantification of 15 IU per milliliter. HCV genotype and subtype were determined with the use of the VERSANT HCV Genotype Assay, version 2 (Siemens) or the TRUGENE HCV 5'NC Genotyping Kit. If the initial assays failed, sequencing of the NS5B regions of HCV RNA was performed.
Assessments during treatment included standard laboratory testing, measurement of HCV RNA levels, symptom-directed physical examinations, and clinical assessments of ascites and encephalopathy. All adverse events were graded according to a standardized scale.
Virologic breakthrough was defined as detectable HCV RNA during treatment in a patient who had previously had undetectable HCV RNA. Relapse was defined as detectable HCV RNA during the post-treatment period in a patient who had undetectable HCV RNA at the end of treatment.
Deep sequencing of the NS5A and NS5B regions was performed on samples obtained from all patients at baseline. For patients who had virologic failure, deep sequencing of the NS5A and NS5B regions was performed on samples obtained at the time of failure. The resulting sequences were compared with baseline sequences to detect emergent variants. Resistance-associated variants that were present in more than 1% of sequence reads were reported.
Study End Points
The primary efficacy end point was the rate of sustained virologic response (defined as HCV RNA <15 IU per milliliter) at 12 weeks after the end of treatment in all patients who underwent randomization and received at least one dose of a study drug. Secondary efficacy end points included the change from baseline in the CPT and MELD scores at 12 weeks after the end of treatment.
Study Oversight
This study was approved by the institutional review board or independent ethics committee at each participating site and was conducted in compliance with the Declaration of Helsinki, Good Clinical Practice guidelines, and local regulatory requirements. The study was designed and conducted according to the protocol by the sponsor (Gilead Sciences) in collaboration with the principal investigators. The sponsor collected the data, monitored study conduct, and performed the statistical analyses. An independent data and safety monitoring committee reviewed the progress of the study. All authors had access to the data and assume responsibility for the integrity and completeness of the reported data. The initial draft of the manuscript was prepared by a professional writer employed by Gilead Sciences and the primary investigators with input from all the authors. All the authors vouch for the fidelity of the study to the protocol.
Statistical Analysis
In the primary efficacy hypothesis, we compared the rate of sustained virologic response in each of the three treatment groups with an assumed spontaneous rate of 1%, using the two-sided exact one-sample binomial test with Bonferroni alpha adjustment. We determined that the enrollment of 75 patients in each treatment group would provide a power of 99% to detect an improvement of at least 40 percentage points in the rate of sustained virologic response over the assumed spontaneous rate of 1%, using the two-sided exact one-sample binomial test at a significance level of 0.0167. We calculated point estimates and two-sided 95% exact confidence intervals that are based on the Clopper-Pearson method for rates of sustained virologic response for the three treatment groups combined, as well as according to HCV genotype and subgroups. The study was not designed or powered to detect significant differences in rates of sustained virologic response among the treatment groups. However, we performed a post hoc pairwise comparison of rates of sustained virologic response among the three treatment groups, for which we calculated point estimates, corresponding 98.3% confidence intervals (rather than 95%, owing to three pairwise comparisons), and P values (using the Cochran-Mantel-Haenszel test). We also performed prespecified analyses of changes in CPT and MELD scores from baseline to post-treatment week 12. The analysis of the change in the MELD score was performed separately for patients with a baseline score of less than 15 and those with a baseline score of 15 or more.
Results
Baseline Characteristics
Of the 438 patients who were screened, 268 underwent randomization and 267 began treatment (Table S1 and Fig. S1 in the Supplementary Appendix, available at NEJM.org). One patient, who was assigned to receive sofosbuvir-velpatasvir plus ribavirin, did not begin treatment because of an adverse event and was omitted from all analyses.
The demographic and baseline clinical characteristics of the patients were generally well balanced (Table 1). Overall, 60% of patients had HCV genotype 1a, 18% genotype 1b, 4% genotype 2, 15% genotype 3, 3% genotype 4, and less than 1% genotype 6; no patients had genotype 5. A total of 6% of patients were black, and 55% had received prior treatment for HCV infection. The median baseline CPT score was 8 (range, 5 to 10), the median baseline MELD score was 10 (range, 6 to 24), and the median creatinine clearance was 84.7 ml per minute (range, 15 to 198). The majority of patients (95%) had a baseline MELD score of 15 or less. All the patients had CPT class B cirrhosis at screening, but 27 patients (10%) had CPT class A or CPT class C cirrhosis at treatment baseline, which reflects the dynamic changes in CPT scoring in this population.
Efficacy
Sustained Virologic Response
Rates of sustained virologic response were 83% (95% confidence interval [CI], 74 to 90) in patients who received sofosbuvir-velpatasvir for 12 weeks, 94% (95% CI, 87 to 98) among those who received sofosbuvir-velpatasvir plus ribavirin, and 86% (95% CI, 77 to 92) among those who received sofosbuvir-velpatasvir for 24 weeks (Table 2). Thus, all three treatment groups met the prespecified primary efficacy end point, with rates of sustained virologic response that were significantly superior to the assumed spontaneous rate of HCV clearance of 1% at 12 weeks after treatment (P<0.001 for all three comparisons). Post hoc analyses did not detect any significant differences in rates of sustained virologic response among the three treatment groups (Table S2 in the Supplementary Appendix).
Among patients with HCV genotype 1, the rate of sustained virologic response was 88% for those who received sofosbuvir-velpatasvir for 12 weeks, 96% for those who received sofosbuvir-velpatasvir plus ribavirin, and 92% for those who received sofosbuvir-velpatasvir for 24 weeks. Among the smaller population of patients with HCV genotype 3, the rate of sustained virologic response among patients who received sofosbuvir-velpatasvir plus ribavirin was 85%, as compared with 50% for the two groups that received sofosbuvir-velpatasvir alone. All the patients with HCV genotype 2, 4, or 6 had a sustained virologic response except for one patient with HCV genotype 2 who was assigned to receive sofosbuvir-velpatasvir for 24 weeks; this patient died of liver failure after completing 28 days of treatment.
A total of 22 patients had virologic failure: 11 of 90 patients (12%) who received sofosbuvir-velpatasvir for 12 weeks, 3 of 87 patients (3%) who received sofosbuvir-velpatasvir plus ribavirin, and 8 of 90 patients (9%) who received sofosbuvir-velpatasvir for 24 weeks. Of the 22 patients who had virologic failure, 20 had a relapse and 2 (both with HCV genotype 3) had virologic breakthrough. One of the patients with virologic breakthrough, a 56-year-old white man who was assigned to receive sofosbuvir-velpatasvir plus ribavirin, had undetectable plasma levels of study drugs at the time of virologic failure, which suggests nonadherence. The other patient with virologic breakthrough was a 52-year-old white man with HCV genotype 3a who was assigned to receive sofosbuvir-velpatasvir for 24 weeks. This patient had an HCV RNA level of less than 15 IU per milliliter from week 4 through week 10 with low levels of HCV RNA (26 to 80 IU per milliliter) at week 12 and week 16; the patient's participation in the study was terminated early at week 16 because he met the stopping criteria for virologic failure. There was no evidence to suggest nonadherence. Characteristics of the patients with virologic failure are provided in Table S3 in the Supplementary Appendix. Also counted among the patients with treatment failure were 4 who were lost to follow-up and 7 who died before the primary end point.
Changes in Liver Function
Of the 267 patients who were treated, 250 had CPT and MELD scores that were available at post-treatment week 12. Of these 250 patients, 117 (47%) had an improvement in the CPT score over baseline, 106 (42%) had no change in the CPT score, and 27 (11%) had a worsening in the CPT score (Figure 1A).
Of the 223 patients with a baseline MELD score of less than 15 for whom MELD data were available at post-treatment week 12, a total of 114 (51%) had an improved MELD score, 49 (22%) had no change in the MELD score, and 60 (27%) had a worsening in the MELD score (Figure 1B). Of the 27 patients who had a baseline MELD score of 15 or more, 22 (81%) had an improved MELD score, 3 (11%) had no change in the MELD score, and 2 (7%) had a worsening in the MELD score (Figure 1C).
Viral Resistance Testing
Of the 255 patients for whom pretreatment NS5A sequencing data were available, 72 (28%) had pretreatment NS5A resistance-associated variants. Of these 72 patients, 64 (89%) had a sustained virologic response, as compared with 169 of 183 patients (92%) who did not have pretreatment NS5A resistance-associated variants.
Among patients with HCV genotype 1 receiving sofosbuvir-velpatasvir plus ribavirin, the rate of sustained virologic response in those with NS5A resistance-associated variants was 100%, and the rate without such variants was 98%. Among patients with HCV genotype 1 in the sofosbuvir-velpatasvir groups who had pretreatment resistance-associated variants, the rate of sustained virologic response was 80% among those who received 12 weeks of treatment and 90% among those who received 24 weeks of treatment; among those who did not have resistance-associated variants, the rates were 96% and 98%, respectively. An analysis of the effect of resistance on treatment outcome in patients with HCV genotype 3 was limited by the small number (6 patients) with resistance-associated variants in our study. The majority of patients who had virologic failure had NS5A resistance-associated variants at the time of failure; NS5B resistance-associated variants were less common and typically observed at low levels (Table S3 in the Supplementary Appendix).
Of the 251 patients for whom pretreatment NS5B deep-sequencing data were available, 8 had pretreatment resistance-associated variants (at positions N142T, L159F, E237G, and M289I). All 8 patients had a sustained virologic response.
Safety
A total of 9 patients discontinued study treatment prematurely because of an adverse event: 1 of 90 patients (1%) who received sofosbuvir-velpatasvir for 12 weeks, 4 of 87 patients (5%) who received sofosbuvir-velpatasvir and ribavirin, and 4 of 90 patients (4%) who received sofosbuvir-velpatasvir for 24 weeks (Table 3). No adverse event that led to discontinuation of a study drug was reported in more than 1 patient. Serious adverse events occurred in 19% of patients who received sofosbuvir-velpatasvir for 12 weeks, 16% of those who received sofosbuvir-velpatasvir plus ribavirin, and 18% of those who received sofosbuvir-velpatasvir for 24 weeks. The most common serious adverse events were hepatic encephalopathy and sepsis (with each event occurring in 5 patients across groups) (Table S5 in the Supplementary Appendix).
The most common adverse events in all groups were fatigue (29%), nausea (23%), and headache (22%), although anemia, diarrhea, and insomnia were also common among the patients who received sofosbuvir-velpatasvir plus ribavirin. Overall, 81% of patients in the groups who received sofosbuvir-velpatasvir alone had at least one adverse event, as compared with 91% of patients receiving sofosbuvir-velpatasvir plus ribavirin (Table 3 and Table S4 in the Supplementary Appendix).
Nine deaths occurred during the study. Two patients died after discontinuing study treatment but within 30 days after the end of treatment, and seven patients died more than 30 days after the end of treatment. Most of the deaths were due to complications of end-stage liver disease (i.e., liver failure, sepsis, or multiorgan failure). The nine deaths were evenly divided among the three treatment groups; none were considered to be related to therapy by the investigator (Table S6 in the Supplementary Appendix).
Reductions in hemoglobin, lymphocytes, and platelets were common in all three groups (Table 3). In the group that received sofosbuvir-velpatasvir plus ribavirin, decreases in hemoglobin to less than 10.0 g per deciliter occurred in 23% of patients and decreases to less than 8.5 g per deciliter in 7% of patients. In the groups that received sofosbuvir-velpatasvir, the rates of decrease in hemoglobin were 8% and 1%, respectively, among those who received 12 weeks and 9% and 1% among those who received 24 weeks. Anemia or reductions in hemoglobin were successfully managed in the majority of patients with a modification of or interruption in the ribavirin dose, although 1 patient was treated with erythropoietin. Two patients who received sofosbuvir-velpatasvir for 12 weeks required the infusion of packed red cells for the treatment of gastrointestinal bleeding. Hyperbilirubinemia that was consistent with hemolysis was primarily observed in patients receiving sofosbuvir-velpatasvir plus ribavirin. Serial median levels of selected safety measurements are provided in Figure S2 in the Supplementary Appendix.
Discussion
In this multicenter, randomized, phase 3 study, patients with decompensated cirrhosis caused by chronic HCV infection had high rates of sustained virologic response in all three treatment groups. As in other studies with combinations of direct-acting antiviral agents, patients with HCV genotype 3 had lower rates of sustained virologic response than did patients with other HCV genotypes. Even so, the rate of 85% among patients with HCV genotype 3 who received sofosbuvir-velpatasvir plus ribavirin was higher than the 71% response rate previously reported among patients with HCV genotype 3 and decompensated cirrhosis.8 In our study, only 50% of patients with genotype 3 who received sofosbuvir-velpatasvir alone had a sustained virologic response, which suggests that the contribution of ribavirin may be particularly important in patients with HCV genotype 3.
Early improvements in hepatic function, as indicated by reductions in CPT and MELD scores, were seen in a substantial proportion of patients, largely due to decreases in bilirubin and increases in albumin levels. Patients in the small subgroup with MELD scores of 15 or more at baseline were most likely to have improvements in MELD scores. Whether these early changes will persist remains to be seen. Patients who had a sustained virologic response were eligible to enroll in a registry study (ClinicalTrials.gov number, NCT01457755), which is designed to evaluate the durability of sustained virologic response and the progression or regression of liver disease for up to 5 years after the end of treatment.
Our results are in line with those of other recent trials of direct-acting antiviral regimens. In the SOLAR-1 and SOLAR-2 trials, patients with genotype 1 or 4 and CPT class B or C cirrhosis who received ledipasvir-sofosbuvir plus ribavirin for 12 or 24 weeks had rates of sustained virologic response of 86 to 89% at 12 weeks after the end of treatment.4,5 In the phase 3 ALLY-1 trial, 12 weeks of treatment with daclatasvir, sofosbuvir, and ribavirin resulted in a sustained virologic response rate of 82% in patients with HCV genotype 1 and CPT class A, B, or C cirrhosis.8
Overall, the rate of adverse events was similar among the three treatment groups. Patients who received sofosbuvir-velpatasvir plus ribavirin had a slightly higher rate of adverse events overall and a substantially higher rate of certain events known to be associated with ribavirin therapy than those who received sofosbuvir-velpatasvir alone. Close monitoring of patients with decompensated liver disease who are receiving a combination of direct-acting antiviral agents remains important in light of the limited safety information concerning these new drugs in this sicker population.22,23
Our study has several limitations. It was not powered to detect significant differences among the three treatment groups. We enrolled only patients with moderate hepatic decompensation, so our results cannot be generalized to patients with more severe liver disease. Also, the numbers of patients with HCV genotype 2, 4, or 6 were too small to allow any conclusions to be drawn about the efficacy of sofosbuvir-velpatasvir among patients with these genotypes. Enrollment was limited to the United States, which may account for the high proportion of patients with HCV genotype 1.
In conclusion, treatment with the fixed-dose combination tablet of sofosbuvir-velpatasvir with or without ribavirin for 12 weeks and with sofosbuvir-velpatasvir for 24 weeks resulted in a high rate of sustained virologic response and early improvements in hepatic function in patients with decompensated cirrhosis caused by HCV of all genotypes. Whether the early improvements in liver function translate to long-term clinical benefits remains to be determined, and future trials are required to assess this regimen in patients with more severe degrees of liver decompensation.
|
| |
|
|
|
|
|