| |
Ledipasvir and sofosbuvir fixed-dose combination with and without ribavirin for 12 weeks in treatment-naive and previously treated Japanese patients with genotype 1 hepatitis C: an open-label, randomised, phase 3 trial
|
| |
| |
Download the PDF here
Download the PDF here
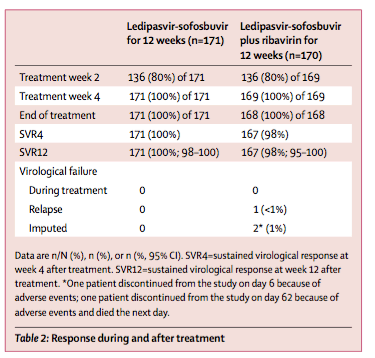
In this trial, 12 weeks of treatment with the fixed-dose combination of ledipasvir and sofosbuvir without ribavirin was well tolerated and resulted in SVR12 in all 171 patients (100%) treated, including patients typically difficult to treat, including those with cirrhosis, or baseline NS5A resistant variants, and those who had previously not responded well to other hepatitis C virus treatment regimens, including protease inhibitor-based therapies. In the ledipasvir-sofosbuvir plus ribavirin group, 167 (98%) of 170 patients achieved SVR12. The addition of ribavirin to ledipasvir-sofosbuvir did not increase the number of patients who achieved SVR12, but did increase the number of patients who had adverse events, even though ribavirin dosing was lower in this study, in accordance with Copegus dosing recommendations in Japan, than in other studies. These findings are broadly in keeping with results from the phase 3 registrational trials of ledipasvir-sofosbuvir in patients with genotype 1 hepatitis C virus done in the USA and Europe.13, 14, 15 In those trials, 12 weeks of ledipasvir-sofosbuvir resulted in 99% of treatment-naive patients and 94% previously treated patients achieving SVR12. Similarly to the present trial, patients receiving ribavirin had similar rates of sustained virological response, but increased rates of adverse events. A previous phase 1 study16 showed that no clinically significant differences existed in the pharmacokinetics of sofosbuvir, GS-331007, or ledipasvir between Japanese and white patients.
Between Oct 15, 2013 and Dec 13, 2013, 341 patients were randomly assigned to treatment groups and received at least one dose of study treatment. SVR12 was achieved in all 171 (100%) patients (83 of 83 treatment naive and 88 of 88 treatment experienced) receiving ledipasvir-sofosbuvir (95% CI 98-100) and 167 (98%) of 170 patients (80 of 83 treatment naive and 87 of 87 treatment experienced) receiving ledipasvir-sofosbuvir plus ribavirin (95% CI 95-100). Of the 76 patients with baseline NS5A resistant variants, 75 (99%) achieved SVR12. Two (1·2%) of 170 patients in the ledipasvir-sofosbuvir plus ribavirin group discontinued treatment because of adverse events. The most common adverse events were nasopharyngitis (50 [29·2%] of 171), headache (12 [7·0%] of 171), and malaise (nine [5·3%] of 171) in patients receiving ledipasvir-sofosbuvir; and nasopharyngitis (40 [23·5%] of 170), anaemia (23 [13·5%] of 170), and headache in those receiving ledipasvir-sofosbuvir and ribavirin (15 [8·8%] of 170).
Overall, most patients (58%) were female and almost all patients (97%) had genotype 1b hepatitis C virus (table 1). The mean age was 59 years, and 112 (33%) patients were aged 65 years or older. 76 patients (22%) had compensated cirrhosis at baseline. Overall, 76 (22%) patients had hepatitis C virus NS5A resistance-associated variants at baseline, 42 (25%) of the 171 receiving ledipasvir-sofosbuvir and 34 (20%) of the 170 receiving ledipasvir-sofosbuvir plus ribavirin. Of the 175 previously treated patients, 40 (23%) had received previous treatment with a triple therapy regimen consisting of pegylated interferon alfa and ribavirin plus a protease inhibitor, such as telaprevir (14), simeprevir (13), vaniprevir (eight), or faldaprevir (five). 88 (50%) of the previously treated patients had relapse or breakthrough during previous hepatitis C virus treatment, 57 (33%) had no response, and 30 (17%) were interferon intolerant.
Overall, 338 (99%) of 341 patients achieved SVR12, including all 171 patients (100%) receiving ledipasvir-sofosbuvir (95% CI 98-100) and 167 (98%) of the 170 patients receiving ledipasvir-sofosbuvir plus ribavirin (95% CI 95-100; table 2). In treatment-naive patients without cirrhosis, 70 (100%) of 70 patients receiving ledipasvir-sofosbuvir and 69 (97%) of 71 patients receiving ledipasvir-sofosbuvir plus ribavirin achieved SVR12, thereby meeting the primary efficacy endpoint of SVR12 better than the prespecified adjusted historical sustained virological response null rate of 63% (p<0.0001 for both comparisons). There was 100% concordance between SVR4, SVR12, and SVR24. Adherence to study drugs was high in both groups (appendix).
----------------
Ledipasvir and sofosbuvir fixed-dose combination with and without ribavirin for 12 weeks in treatment-naive and previously treated Japanese patients with genotype 1 hepatitis C: an open-label, randomised, phase 3 trial
Lancet Infect Dis June 2015
Masashi Mizokami, Osamu Yokosuka, Tetsuo Takehara, Naoya Sakamoto, Masaaki Korenaga, Hitoshi Mochizuki, Kunio Nakane, Hirayuki Enomoto, Fusao Ikeda, Mikio Yanase, Hidenori Toyoda, Takuya Genda, Takeji Umemura, Hiroshi Yatsuhashi, Tatsuya Ide, Nobuo Toda, Kazushige Nirei, Yoshiyuki Ueno, Yoichi Nishigaki, Juan Betular, Bing Gao, Akinobu Ishizaki, Masa Omote, Hongmei Mo, Kim Garrison,Phillip S Pang, Steven J Knox, William T Symonds, John G McHutchison, Namiki Izumi, Masao Omata
Summary
Background
Compared with other countries, patients with chronic hepatitis C infection in Japan tend to be older, have more advanced liver disease, and are more likely to have been previously treated for hepatitis C. We aimed to assess the efficacy and safety of an all-oral, fixed-dose combination of the hepatitis C virus NS5A inhibitor ledipasvir and the NS5B nucleotide polymerase inhibitor sofosbuvir with and without ribavirin for 12 weeks in treatment-naive and previously treated Japanese patients with chronic genotype 1 hepatitis C virus infection.
Methods
In this randomised, open-label study, we enrolled patients from 19 clinical Japanese centres. Patients were randomly assigned (1:1) to receive either ledipasvir (90 mg) and sofosbuvir (400 mg) or ledipasvir, sofosbuvir, and ribavirin (dosed according to the Japanese Copegus product label-ie, patients ≤60 kg received 600 mg daily, patients >60 kg to ≤80 kg received 800 mg daily, and patients >80 kg received 1000 mg daily) orally once daily for 12 weeks. After completion or early discontinuation of treatment, patients were followed up off-treatment for 24 weeks. Eligible patients were at least 20 years of age with chronic genotype 1 hepatitis C virus infection with serum hepatitis C virus RNA concentrations of at least 5 log10 IU/mL, creatinine clearance of at least 1·0 mL/s, and a platelet count of at least 50 x 109 per L. An interactive web response system was used to manage patient randomisation and treatment assignment. Randomisation was stratified by the presence or absence of cirrhosis for treatment-naive patients and stratified by presence or absence of cirrhosis and by previous treatment category (relapser or breakthrough, non-responder, or interferon-intolerant) for previously treated patients. Within each strata, patients were sequentially assigned to either treatment with ledipasvir-sofosbuvir or ledipasvir-sofosbuvir plus ribavirin in a 1:1 ratio with block size of 4. The primary endpoint was sustained virological response 12 weeks after completion of treatment (SVR12) assessed in all patients who were randomly assigned and received at least one dose of study drug; safety outcomes were assessed in all patients who received at least one dose of study drug. This trial is registered with ClinicalTrials.gov, number NCT01975675.
Findings
Between Oct 15, 2013 and Dec 13, 2013, 341 patients were randomly assigned to treatment groups and received at least one dose of study treatment. SVR12 was achieved in all 171 (100%) patients (83 of 83 treatment naive and 88 of 88 treatment experienced) receiving ledipasvir-sofosbuvir (95% CI 98-100) and 167 (98%) of 170 patients (80 of 83 treatment naive and 87 of 87 treatment experienced) receiving ledipasvir-sofosbuvir plus ribavirin (95% CI 95-100). Of the 76 patients with baseline NS5A resistant variants, 75 (99%) achieved SVR12. Two (1·2%) of 170 patients in the ledipasvir-sofosbuvir plus ribavirin group discontinued treatment because of adverse events. The most common adverse events were nasopharyngitis (50 [29·2%] of 171), headache (12 [7·0%] of 171), and malaise (nine [5·3%] of 171) in patients receiving ledipasvir-sofosbuvir; and nasopharyngitis (40 [23·5%] of 170), anaemia (23 [13·5%] of 170), and headache in those receiving ledipasvir-sofosbuvir and ribavirin (15 [8·8%] of 170).
Interpretation
Although existing regimens for the treatment of hepatitis C virus are effective for many patients, medical needs remain unmet, particularly in Japan where the population with hepatitis C virus genotype 1 is generally older and treatment-experienced, with advanced liver disease. The efficacy, tolerability, and absence of drug-drug interactions of ledipasvir-sofosbuvir suggest that it could be an important option for treatment of genotype 1 hepatitis C virus in Japanese patients.
Funding
Gilead Sciences.
Introduction
The prevalence of chronic hepatitis C virus infection in Japan is estimated at up to 2 million people (1·6-2% of the population).1 Of these, roughly 70% have genotype 1 hepatitis C virus, the most common hepatitis C virus strain worldwide and the most difficult to cure with interferon-based treatments. Japanese patients with hepatitis C virus are older (65-76% are >60 years), frequently treatment experienced (with interferon-based therapies), and at high risk of developing hepatocellular carcinoma.1 Guidelines issued by the Japan Society of Hepatology for patients with genotype 1 hepatitis C virus recommend as the first treatment of choice 12 weeks of triple therapy with the hepatitis C virus NS3/4A protease inhibitor simeprevir, pegylated interferon alfa, and ribavirin followed by 12-36 additional weeks of pegylated interferon alfa and ribavirin if the patient is eligible for and tolerant of interferon.2 Although this combination provides high rates of sustained virological response (defined as hepatitis C virus RNA < lower level of quantification [LLOQ] after completion of antiviral therapy for chronic hepatitis C infection) in treatment-naive patients and patients who have previously relapsed who are eligible to receive interferon, sustained virological response in patients with previous non-response to treatment is substantially reduced (36-53%).3, 4, 5 Moreover, the side-effects and drug interactions associated with protease inhibitor regimens are problematic for patients with progressive liver disease and comorbid conditions, and for patients with relative or absolute contraindications to interferon or ribavirin.6, 7 For patients who cannot receive interferon, a regimen of daclatasvir plus asunaprevir for 24 weeks is recommended.2Although this combination provides an alternative interferon-free regimen, this drug combination is only approved for selected patient populations (patients who have Y93 or L31 mutations in the NS5A region of the hepatitis C virus are not recommended to receive this therapy),2 has suboptimum efficacy and resistance profiles,8 and is associated with specific toxic effects.9, 10 An unmet medical need for a simple, safe, and effective regimen that can be used in an ageing population with progressive liver disease remains.
Research in context
Evidence before this study
In January, 2013, we consulted Japanese hepatitis C virus experts and did a multi-institution survey to confirm the demographic and disease characteristics of patients with chronic hepatitis C virus in Japan. Data from this survey suggested that most Japanese patients with hepatitis C virus are elderly (65-76% are >60 years old), more than half are treatment-experienced, and nearly 30% have advanced fibrosis. Fewer than 10% of patients were willing or able to receive interferon. Furthermore, we consulted comprehensive reviews of hepatitis C virus and its treatment in Japan and hepatitis C virus treatment guidelines issued by the Japan Society of Hepatology. Finally, we did PubMed searches for Articles using the terms "HCV treatment" and "Japan" on June 3, 2013 and Nov 25, 2014. There were no language restrictions for this search.
In Japanese patients, telaprevir for 12 weeks with peginterferon and ribavirin for 24 weeks resulted in a sustained virological response rate of 80-87% in treatment-naive patients, 85-88% in previous relapsers, and 75-100% in previous non-responders. In the CONCERTO-4 study, treatment with simeprevir plus peginterferon and ribavirin for 24 weeks led to sustained virological response in 92% of treatment-naive patients, 100% of previous relapsers, and 39% of previous non-responders. In a phase 2 study, a sustained virological response rate of 95-100% was reported in treatment-experienced Japanese patients who received vaniprevir (100 mg, 300 mg, or 600 mg) for 4 weeks with peginterferon plus ribavirin for 6-72 weeks. Safety and tolerability of these regimens is limited by the need for concomitant interferon and ribavirin combined with adverse events and laboratory abnormalities associated with the hepatitis C virus NS3/4A protease inhibitor.
Recently, the all-oral, 24-week regimen of daclatasvir (NS5A inhibitor) plus asunaprevir (NS3A protease inhibitor) was approved in Japan for specific populations. In a phase 3 trial, SVR12 was achieved by 88% of treatment-naive, interferon-ineligible patients and 81% of previous non-responders. Importantly, of those who did not achieve sustained virological response, 65% had hepatitis C virus NS5A resistance-associated variants (L31M/V or Y93H) at baseline and 85% had resistance-associated variants to both daclatasvir and asunaprevir at the time of virological failure. Japanese treatment guidelines recommend use of this regimen in a limited population and require assessment of resistance-associated variants before initiating therapy
Added value of this study
In this study, we deliberately included patients who have been under-represented in trials-elderly patients and those who cannot receive interferon-as well as patients with characteristics that have been associated with reduced sustained virological response rates-patients with resistance-associated variants at baseline, cirrhosis, and treatment-experienced patients. Treatment with the single-tablet regimen of ledipasvir and sofosbuvir taken once daily for 12 weeks was well tolerated and cured all patients treated. The absence of drug interactions means that it could be used irrespective of concomitant medical conditions and treatment.
Implications of all the available evidence
Although existing regimens for the treatment of hepatitis C virus are effective for many patients, important medical needs remain unmet, particularly in Japan where the population with hepatitis C virus genotype 1 is generally older and treatment-experienced, with advanced liver disease. The efficacy, tolerability, and absence of drug-drug interactions of ledipasvir-sofosbuvir suggest that it could be an important option for treatment of genotype 1 hepatitis C virus in a broad range of Japanese patients.
Sofosbuvir is a nucleotide analogue inhibitor of the hepatitis C virus non-structural protein 5B (NS5B) polymerase that is approved in the USA, Europe, and other countries for the treatment of patients with chronic hepatitis C virus infection.11 Ledipasvir is a novel hepatitis C virus NS5A inhibitor that has shown potent anti-hepatitis C virus activity.12 In three phase 3 trials done in the USA and Europe, 12 weeks of treatment with the ledipasvir-sofosbuvir fixed-dose combination was well tolerated and resulted in high rates (94-99%) of sustained virological response in treatment-naive and previously treated patients with genotype 1 hepatitis C virus, including those with cirrhosis.13, 14, 15 Subsequently, ledipasvir-sofosbuvir received marketing authorisation in these regions in 2014.
We did a phase 3 trial to assess the efficacy and safety of 12 weeks of the ledipasvir-sofosbuvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated Japanese patients with genotype 1 hepatitis C virus infection. The primary efficacy endpoint of this study was sustained virological response 12 weeks after the end of treatment (SVR12).
Methods
Study design and participants
In this randomised, open-label, phase 3 trial, we enrolled patients with chronic genotype 1 hepatitis C virus infection at 19 clinical sites in Japan. Planned patient enrolment was 150 treatment-naive and 150 treatment-experienced patients (appendix). Eligible patients were at least 20 years of age with chronic genotype 1 hepatitis C virus infection with serum hepatitis C virus RNA concentrations of at least 5 log10 IU/mL and creatinine clearance of at least 1·0 mL/s (Cockcroft-Gault equation). We did not use an upper age limit. Patients with hepatic decompensation (as shown by the presence of ascites, encephalopathy, or a history of variceal haemorrhage), bodyweight less than 40 kg, or coinfection with hepatitis B or HIV were excluded. Consistent with a population with progressive liver disease, no minimum neutrophil count was needed and patients with a platelet count of at least 50 x 109 platelets per L were eligible for participation. Up to 40% of patients enrolled in the study could have had compensated cirrhosis. The presence of cirrhosis was established either by liver biopsy (eg, a Metavir score of 4 or an Ishak score of ≥5) or by Fibroscan score of more than 12·5 kPa. All patients were eligible to participate in a substudy to establish the steady-state pharmacokinetics of sofosbuvir (and its metabolite GS-331007), ledipasvir, and, where applicable, ribavirin.
Before enrolment and before any study procedures were undertaken, the study was approved by appropriate regulatory bodies and written informed consent was obtained from all patients. This study was done in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice. See appendix for protocol.
Randomisation and masking
Patients were randomly assigned (1:1) to receive either ledipasvir-sofosbuvir or ledipasvir-sofosbuvir and ribavirin. An interactive web response system was used to manage patient randomisation and treatment assignment. Randomisation of treatment-naive patients was stratified by the presence or absence of cirrhosis. Randomisation of previously treated patients was stratified by presence or absence of cirrhosis and by previous treatment category: relapser or breakthrough, non-responder, or interferon-intolerant. Within each strata, patients were sequentially assigned to treatments ledipasvir-sofosbuvir or ledipasvir-sofosbuvir plus ribavirin in a 1:1 ratio with a block size of 4. This study used an open-label design; both patients and study investigators were aware of treatment assignment.
Procedures
Patients in both treatment groups received 12 weeks of treatment with a fixed-dose combination tablet containing 90 mg of ledipasvir and 400 mg of sofosbuvir (ledipasvir-sofosbuvir) orally, once daily with or without food. When assigned, ribavirin was given orally as a divided, weight-based daily dose according to the Japanese Copegus product label (ie, patients ≤60 kg received 600 mg daily, patients >60 kg to ≤80 kg received 800 mg daily, and patients >80 kg received 1000 mg daily). After completion or early discontinuation of treatment, patients were followed up off-treatment for 24 weeks.
Serum hepatitis C virus RNA concentrations were measured with the COBAS TaqMan hepatitis C virus Test, version 2·0 for use with the High Pure System (Roche, IN, USA), with a lower limit of quantification for hepatitis C virus RNA of less than 25 IU/mL. Virological breakthrough was defined as the presence during treatment of hepatitis C virus RNA of at least 25 IU/mL in a patient with previous documentation of on-treatment concentrations of hepatitis C virus RNA of less than 25 IU/mL. Relapse was defined as the confirmed presence of hepatitis C virus RNA of at least 25 IU/mL at any time during the post-treatment follow-up after documented hepatitis C virus RNA of less than 25 IU/mL at the end of treatment. For all patients, the IL28B genotype was established by PCR amplification and sequencing of the rs12979860 single nucleotide polymorphism.
Deep sequencing of the NS5A and NS5B regions of the hepatitis C virus was done for all patients at baseline. For all patients who had virological failure (defined as virological breakthrough, relapse, rebound [>1 log10 IU/mL increase in hepatitis C virus RNA from nadir while on treatment with two consecutive values, or last available on-treatment measurement with no subsequent follow up values], or non-response [hepatitis C virus RNA persistently >25 IU/mL through 8 weeks of treatment]), deep sequencing of the NS5A and NS5B regions was done at both baseline and at the time of virological failure. The resulting sequences were compared with sequences from baseline samples to detect treatment emergent resistance-associated variants. Resistance-associated variants present at greater than 1% of sequence reads were regarded as significant.
The population pharmacokinetic variables for ledipasvir, sofosbuvir, and GS-331007 (the predominant circulating metabolite of sofosbuvir) were computed for all patients from available concentration data from intensive or sparse samples with previously established population pharmacokinetic models.
Outcomes
The primary efficacy endpoint of the study was the proportion of patients who achieved SVR12. Analysis of this endpoint included all patients who were randomly assigned and received at least one dose of study drug. SVR12 is the proportion of patients with hepatitis C virus RNA less than LLOQ (ie <25 IU/mL) 12 weeks after completion of treatment. Secondary outcomes were to establish the proportion of patients who attained sustained virological response at 4 and 24 weeks after discontinuation of therapy (SVR4 and SVR24), to assess the kinetics of circulating HCV RNA during treatment and after treatment discontinuation, and to assess the emergence of viral resistance to sofosbuvir and ledipasvir during treatment and after treatment discontinuation. Safety was assessed in all patients by physical examination and by review of adverse events and blood and urine samples for clinical laboratory testing. Reduction or discontinuation of ribavirin dosing because of toxic effects was done according to the Japanese Copegus product label. Patients were not permitted to use erythropoiesis-stimulating drugs, granulocyte colony-stimulating factor, or thrombopoietin mimetics from 28 days before screening to the end of treatment.
Statistical analysis
We calculated the SVR12 for each treatment group along with the two-sided 95% CI using the exact binomial distribution (the Clopper-Pearson method). For treatment-naive patients without cirrhosis, the SVR12 rate was compared with the adjusted historical sustained virological response null rate of 63% using a two-sided exact one-sample binomial test. The appendix shows the method of calculating the historical sustained virological response control rate. No statistical hypothesis testing was done for outcomes in treatment-naive patients with cirrhosis or in previously treated patients.
We estimated that a sample size of 45 treatment-naive patients without cirrhosis will provide at least 90% power to detect a 23% improvement in SVR12 rate from the adjusted historical control rate of 63% using a two-sided exact one-sample binomial test at a significance level of 0.025 based on a Bonferroni correction. The targeted enrolment for the pharmacokinetics substudy was roughly 15 each of treatment-naive and previously treated patients. SAS Software version 9.2 was used for statistical analysis (SAS Institute, NC, USA).
This trial is registered with ClinicalTrials.gov, number NCT01975675.
Role of the funding source
The funder of the study oversaw trial management, data collection, statistical analyses, and the writing and review of the report. The corresponding author had full access to all data in the study and had final responsibility for the decision to submit for publication.
Results
Screening for the trial began on Oct 15, 2013, with the last patient enrolled on Dec 13, 2013. 421 patients were screened, of whom all 341 were randomly assigned and received study treatment (figure; appendix). Overall, most patients (58%) were female and almost all patients (97%) had genotype 1b hepatitis C virus (table 1). The mean age was 59 years, and 112 (33%) patients were aged 65 years or older. 76 patients (22%) had compensated cirrhosis at baseline. Overall, 76 (22%) patients had hepatitis C virus NS5A resistance-associated variants at baseline, 42 (25%) of the 171 receiving ledipasvir-sofosbuvir and 34 (20%) of the 170 receiving ledipasvir-sofosbuvir plus ribavirin. Of the 175 previously treated patients, 40 (23%) had received previous treatment with a triple therapy regimen consisting of pegylated interferon alfa and ribavirin plus a protease inhibitor, such as telaprevir (14), simeprevir (13), vaniprevir (eight), or faldaprevir (five). 88 (50%) of the previously treated patients had relapse or breakthrough during previous hepatitis C virus treatment, 57 (33%) had no response, and 30 (17%) were interferon intolerant.
Overall, 338 (99%) of 341 patients achieved SVR12, including all 171 patients (100%) receiving ledipasvir-sofosbuvir (95% CI 98-100) and 167 (98%) of the 170 patients receiving ledipasvir-sofosbuvir plus ribavirin (95% CI 95-100; table 2). In treatment-naive patients without cirrhosis, 70 (100%) of 70 patients receiving ledipasvir-sofosbuvir and 69 (97%) of 71 patients receiving ledipasvir-sofosbuvir plus ribavirin achieved SVR12, thereby meeting the primary efficacy endpoint of SVR12 better than the prespecified adjusted historical sustained virological response null rate of 63% (p<0.0001 for both comparisons). There was 100% concordance between SVR4, SVR12, and SVR24. Adherence to study drugs was high in both groups (appendix).
Response in patient subgroups is shown in the appendix. All 175 (100%) patients previously treated achieved SVR12, including all 40 who did not respond to previous treatment with a protease inhibitor-based regimen. In patients with NS5A resistance-associated variants at baseline, 42 (100%) of 42 of those receiving ledipasvir-sofosbuvir and 33 (97%) of 34 receiving ledipasvir-sofosbuvir plus ribavirin achieved SVR12.
In view of the uniformly high levels of SVR12 in all groups, identification of factors predictive of virological success or failure was not possible. Patients with factors that have historically been associated with lower response-non-response to previous treatment, the presence of cirrhosis, non-CC IL28B genotype-had SVR12 rates similar to patients without unfavourable characteristics. The presence of ribavirin in the regimen was not associated with increased SVR12.
In 341 patients enrolled and treated, one had virological failure. This patient, a treatment-naive 55-year-old woman without cirrhosis who was receiving ledipasvir-sofosbuvir plus ribavirin, relapsed by post-treatment week 4 after completion of treatment. This patient had adherence rates of more than 99% for both ledipasvir-sofosbuvir and ribavirin (800 mg daily) based on pill counts from returned study medication.
The patient who had a viral relapse had genotype 1b hepatitis C virus infection and the Y93H (>99%) NS5A resistance-associated variants at baseline and at post-treatment week 4. No other NS5A resistance-associated variants were detected at post-treatment week 4. No NS5B nucleoside inhibitor variants and no treatment-emergent variants were detected in any patient at any timepoint tested.
The mean (coefficient of variation [CV], %) of steady-state for ledipasvir for AUC0-24 (defined as area under the concentration-time curve from 0 h to 24 h) was 11 500 (56·0) ng x h per mL, for Cmax (defined as maximum or peak plasma concentration) was 485 (48·3) ng/mL, and for Ctau (the plasma concentration at the end of the dosing interval) was 326 (55·9) ng/mL. The mean (CV, %) was 1560 (43·1) ng x h per mL for steady-state AUC0-24 and 579 (40·3) ng/mL for Cmax for sofosbuvir (n=67). For its metabolite GS-331007 (n=341), steady-state AUC0-24 was 12 600 (23·9) ng x h per mL, and Cmax was 723 (22·1) ng/mL. We noted no clinically relevant differences in the pharmacokinetics of ledipasvir, sofosbuvir, or GS-331007 on the basis of creatinine clearance, age, sex, body-mass index, cirrhosis status, previous treatment experience, or ribavirin treatment regimen.
Overall, 240 (70%) of 341 patients had at least one treatment-emergent adverse event. Of the patients who had adverse events, most (201 [84%] of 240) had only mild (grade 1) events. The most common adverse events were nasopharyngitis (common cold), anaemia, and headache. Adverse events were higher in patients receiving ledipasvir-sofosbuvir plus ribavirin (128 [75%] of 170) than in those receiving ledipasvir-sofosbuvir only (112 [65%] of 171). Patients receiving ribavirin had higher rates of events known to be associated with this drug-anaemia, pruritus, rash, and nausea. Modification of ribavirin dose was needed in 20 (12%) of 170 patients (appendix), mainly because of anaemia or decreased haemoglobin. Two patients receiving ledipasvir-sofosbuvir plus ribavirin discontinued all study treatment prematurely because of adverse events: one patient discontinued treatment because of a ribavirin-associated morbilliform drug eruption on day 6 of treatment, and one patient had a cardiac arrest leading to death 1 day after stopping treatment. None of the patients randomly assigned to receive ledipasvir-sofosbuvir discontinued the study drugs because of adverse events.
Five patients had a treatment-emergent serious adverse event, of which two were judged to be related to study treatment. One patient, a 67-year-old, treatment-naive man with a medical history including liver cirrhosis, diabetes mellitus, sarcoidosis, pulmonary fibrosis, splenectomy, and chronic gastritis was randomly assigned to receive ledipasvir-sofosbuvir plus ribavirin. On day 62 (week 8) of treatment, the patient had nausea, vomiting, diarrhoea, and fever suggestive of a serious underlying infection; at that time, study treatment was discontinued. The next day, he had a cardiac arrest and died. The investigator assessed the death as related to study treatment, but also listed possible alternate causes, including pre-existing conditions, persisting infection, and other, unspecified, drugs. The investigator commentary reported a low likelihood of association between death and the investigational drugs, with the most probable explanation being viral gastrointestinal infection leading to cardiac arrest.
The other serious adverse event judged by an investigator as related to study treatment was an acute myocardial infarction that occurred in a treatment-experienced, 71-year old man, who had previously received pegylated interferon alfa-2b and ribavirin and had a history of liver cirrhosis and hypertension. This patient received 12 weeks of ledipasvir-sofosbuvir plus ribavirin. 9 days after completion of treatment, the patient had an acute myocardial infarction. The patient underwent cardiac catheterisation and was identified to have complete stenosis of the left anterior descending artery and 99% stenosis of the right coronary artery, which probably represented pre-existing chronic conditions (appendix).
The rate of adverse events in patients younger than 65 years (155 [68%] of 229) was lower than that identified in patients aged 65 years and older (85 [76%] of 112). Overall, patients with cirrhosis did not have a substantially higher rate of adverse events than did patients without cirrhosis (appendix).
12 patients (7%) receiving ledipasvir-sofosbuvir and 14 (8%) receiving ledipasvir-sofosbuvir plus ribavirin had grade 3 laboratory abnormalities and none of the patients had any grade 4 abnormalities (appendix). The only clinically significant difference in the prevalence of these laboratory abnormalities between the treatment groups was that a greater number of patients in the ribavirin-treated group had grade 3 reductions in haemoglobin than patients receiving ledipasvir-sofosbuvir only (five [3%] of 170 vs two [1%] of 171). For patients receiving ribavirin, the mean change in haemoglobin from baseline to the end of treatment week 12 was -16 g/L (SD 11·5) versus -3 g/L (SD 8·2) for those receiving ledipasvir-sofosbuvir only. In patients receiving ledipasvir-sofosbuvir and ribavirin, ten (6%) had at least one post-baseline haemoglobin value lower than 100 g/L and none had a post-baseline value lower than 85 g/L versus four (2%) who had at least one post-baseline haemoglobin value lower than 100 g/L and one (≤1%) who had a post-baseline value lower than 85 g/L in the group receiving ledipasvir-sofosbuvir only.
Discussion
In this trial, 12 weeks of treatment with the fixed-dose combination of ledipasvir and sofosbuvir without ribavirin was well tolerated and resulted in SVR12 in all 171 patients (100%) treated, including patients typically difficult to treat, including those with cirrhosis, or baseline NS5A resistant variants, and those who had previously not responded well to other hepatitis C virus treatment regimens, including protease inhibitor-based therapies. In the ledipasvir-sofosbuvir plus ribavirin group, 167 (98%) of 170 patients achieved SVR12. The addition of ribavirin to ledipasvir-sofosbuvir did not increase the number of patients who achieved SVR12, but did increase the number of patients who had adverse events, even though ribavirin dosing was lower in this study, in accordance with Copegus dosing recommendations in Japan, than in other studies. These findings are broadly in keeping with results from the phase 3 registrational trials of ledipasvir-sofosbuvir in patients with genotype 1 hepatitis C virus done in the USA and Europe.13, 14, 15 In those trials, 12 weeks of ledipasvir-sofosbuvir resulted in 99% of treatment-naive patients and 94% previously treated patients achieving SVR12. Similarly to the present trial, patients receiving ribavirin had similar rates of sustained virological response, but increased rates of adverse events. A previous phase 1 study16 showed that no clinically significant differences existed in the pharmacokinetics of sofosbuvir, GS-331007, or ledipasvir between Japanese and white patients.
Our findings suggest that many of the host and viral factors that are predictive of outcomes with interferon-based treatments-previous treatment response, hepatitis C virus genotype (1a vs 1b), age, baseline viral load, IL28B genotype, cirrhosis status, presence of baseline resistant variants, ribavirin exposure, and early treatment response-are not of clinical relevance in the use of the ledipasvir-sofosbuvir fixed-dose combination. Although most patients in this study were hepatitis C virus genotype 1b, consistent with genotype distribution in Japan,1 the efficacy of ledipasvir-sofosbuvir in hepatitis C virus genotype 1a has been extensively examined in overseas studies and no clinically important differences in response have been identified between subtypes.13, 14, 15 This uniformity of response to the regimen, and its potential for use in patients with contraindications to or intolerance of interferon or ribavirin, suggests that this combination might have broad applications.
Ledipasvir-sofosbuvir with and without ribavirin seemed to be well tolerated by patients with hepatitis C virus infection, with adverse events known to be associated with ribavirin therapy.17 Two serious cardiac adverse events (arrest and myocardial infarction) occurred, which were thought to be possibly related to study drug by the investigator; however, in both cases, alternative causes for these adverse events were also proposed by the investigator as likely contributory (infection and pre-existing disease). Data from the present trial and those previously reported for the ledipasvir-sofosbuvir development programme do not suggest that ledipasvir-sofosbuvir confers any increased risk of cardiac events.
With the exception of nasopharyngitis, the type, frequency, and severity of the adverse events reported in patients receiving ledipasvir-sofosbuvir were similar to those seen in untreated patients from the control group of a phase 3 trial.18 No clinically relevant differences in pharmacokinetic variables between Japanese patients in this trial and the population in large phase 3 trials exist.
The proportion of patients with nasopharyngitis was much higher in this study (22-28%; table 3) than in large phase 3 trials, in which the proportion of patients with nasopharyngitis ranged from 1% or less to 9%. The increased rate of nasopharyngitis seen in this study is perhaps attributable to the fact that this study was done during winter in Japan and the population enrolled in the trial.
Limitations of this study include the open-label design and absence of an active comparator. The trial design could introduce bias in reported adverse events and efficacy results secondary to influence on adherence and tolerability (eg, if patients know that they are assigned to receive ribavirin, whereas others in the study are not, they might be selectively non-adherent to ribavirin or be more likely to report known side-effects of ribavirin). The absence of an active comparator also precluded us from assessing which adverse events resulted from ledipasvir-sofosbuvir.
Our findings suggest that the all-oral, interferon-free and ribavirin-free fixed-dose combination of ledipasvir and sofosbuvir given as one tablet once daily might be an important advancement in the treatment of hepatitis C virus genotype 1 in Japan.
|
|
| |
| |
|
|
|