| |
Review article: the efficacy and safety of daclatasvir in
the treatment of chronic hepatitis C virus infection
|
| |
| |
Download the PDF here
(FDA approval expected Aug 2015)
Bristol-Myers Squibb Announces Global HCV Strategy
........ http://www.natap.org/2014/HCV/110314_03.htm
--------------------------
EASL: BMS at EASL - (05/11/15)....in this link are the phase 3 study results - ALLY-1 (advanced cirrhosis/post-transplant), ALLY-2 (HIV coinfection), ALLY-3 (genotype 3), as well as the Compassionate Use Programs by UK, France, BMS, and the cohort studies
(HIV DART/Dec-2014) - Daclatasvir: Overview of Drug-Drug Interactions With Antiretroviral Agents and Other Common Concomitant Drugs.......http://www.natap.org/2014/HCV/121714_01.htm
HCV Drug Interactions & Switching Patient's ART Regimen in order to take HCV Therapy - Clin Pharm Workshop......http://www.natap.org/2015/HCV/061515_12.htm
CROI/2012: Assessment of HIV Antiretroviral Drug Interactions With the HCV NS5A Replication Complex Inhibitor Daclatasvir Demonstrates a PK Profile Which Supports Coadministration With Tenofovir, Efavirenz and Atazanavir/r http://www.natap.org/2012/CROI/croi_38.htm
---------------------------
Review article: the efficacy and safety of daclatasvir in the treatment of chronic hepatitis C virus infection
Alimentary Pharmacology & Therapeutics Aug 2015
C. Bunchorntavakul*, & K. R. Reddy
*Division of Gastroenterology and Hepatology, Department of Medicine, Rajavithi Hospital, College of Medicine, Rangsit University, Bangkok, Thailand.
Division of Gastroenterology and Hepatology, Department of Medicine, University of Pennsylvania, Philadelphia, PA, USA.
(FDA approval expected Aug 2015)
Through evidence from multiple clinical trials, DCV has been approved by two stringent regulatory authorities to date. In Japan, DCV was approved as a dual combination of DCV and ASV in July 2014, bringing to Japanese patients with HCV the first all-oral, IFN-/RBV-free treatment regimen. Subsequently in August 2014, DCV was approved by the European Commission (EC) and the European Medicines Agency for use as combination therapy with other agents across GTs 1, 2, 3 and 4 for the treatment of HCV. These approvals led to the marketing of DCV (Daklinza; Bristol-Myers Squibb, Inc., Princeton, NJ, USA) in Japan and European countries. In the USA, DCV is not FDA approved and thus the use of it is limited to clinical trials and compassionate use programs.
The new HCV treatment paradigm clearly aims towards all-oral regimens, preferably with pangenotypic activity and not more than 12 weeks treatment duration, and with >95% success rates. DCV has relatively low barrier to resistance so that additional potent DAA is (are) required to achieve this goal.
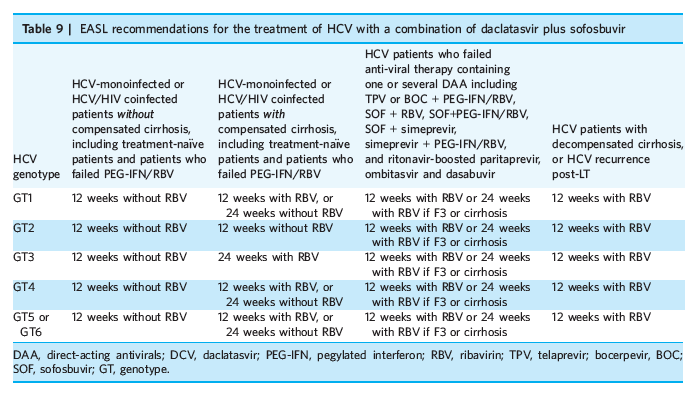
Phase II and III studies have shown that DCV works very well in combination with SOF and currently is one of the best available, once-daily, all-oral regimen, and with multiple genotypic activity (SVR 93-95% for GT1 and 86-90% for GT3).[53] The 2015 EASL Guideline has recommended the use of this combination as an option for various HCV GT and patient populations (Table 9).[53] In Phase III studies, a twice-daily single-pill triple combination of DCV, ASV and BCV, so-called BMS-TRIO, demonstrated excellent SVR rates against GT1b and GT4, but again with slightly decreased SVR rates for GT1a (85-90%). Additional information on the BMS-TRIO, as well as other DCV-containing regimens, are eagerly awaited.
Summary
Daclatasvir is the first-in-class HCV NS5A inhibitor which has shown significant promise in clinical trials. It has good anti-viral potency against multiple HCV GTs, but with low barrier to resistance, which can be overcome by adding PEG-IFN and/or other DAAs to the regimens. DCV is generally safe and well tolerated with mild potential for DDI. When DCV is added to PEG-IFN/RBV, SVR rates are increased (approximately up to 60-100% in GT1,[14-16] 80% in GT2,[23] 70% in GT3,[23] and 82% in GT4[31]), from PEG-IFN/RBV alone. The all-oral combination DCV and ASV has high SVR rates against GT1b (80-95%)[24, 32-34] leading to its approval in Japan, but has less activity against GT1a (23-40% in experienced patients)[21, 22] making it less than an ideal combination for GT1a-predominant areas, such as the US and many countries in Europe. Dual combination of DCV/SOF and triple combination and DCV/ASV/BCV have demonstrated >90% SVR rates in patients with GT1, both naïve and treatment-experienced. Furthermore, DCV/SOF combination has also demonstrated up to 90% SVR rates in patients with GT3, and DCV/ASV/BCV has primarily demonstrated near 100% SVR rates in patients with GT4. High SVR rates in Phase III studies in special populations, such as those with decompensated cirrhosis, HIV coinfection and LT recipients, supports the use of these regimens in patients with a chronic HCV and with multiple GTs.
Daclatasvir and SOF, with and without RBV, as combination therapy has demonstrated impressive efficacy in a large Phase II study by Sulkowski et al.[26] This study initially randomly assigned 44 previously untreated patients with GT1 and 44 patients with GT2/3 to DCV plus SOF, with or without RBV, for 24 weeks. Subsequently, the study was expanded to include 123 additional patients with GT1 to be treated with DCV plus SOF, with or without RBV, for 12 weeks (82 naïve patients) or 24 weeks (41 patients who had previous failed TPV or BOC plus PEG-IFN/RBV).[26] All naïve GT1 patients in the 24-week treatment cohorts achieved SVR24 while GT1 patients in the 12 week groups achieved SVR24 at rates of 93-95%.[26] The SVR rates were 88-100% in naïve patients with GT2/3 and were 95-100% in patients with GT1 who previously failed protease inhibitors.[26] Notably, SVR rates were not related to IL28B status, GT subtype or the administration of RBV.[26]
Sulkowski study -
EASL/2012: Potent Viral Suppression With the All-Oral Combination of Daclatasvir (NS5A Inhibitor) and GS-7977 (Nucleotide NS5B Inhibitor), +/- Ribavirin, in Treatment-Naive Patients With Chronic HCV GT1, 2, or 3 (100% SVR gt1, 91% gt2)- (04/19/12)
EASL: Sustained Virologic Response With Daclatasvir Plus Sofosbuvir ± Ribavirin (RBV) in Chronic HCV Genotype (GT) 1-Infected Patients Who Previously Failed Telaprevir (TVR) or Boceprevir (BOC)- (04/27/13)
Due to the impressive results of DCV/SOF combination for GT1 and GT3 in Phase II study,[26] this dual combination was again evaluated without RBV in a Phase III study (ALLY-3) which included 101 treatment-naïve and 51 treatment-experienced patients with HCV GT3.[35] All patients received a 12-week regimen of once daily DCV 60 mg plus SOF 400 mg, which resulted in SVR12 in 90% of treatment-naïve and 86% of treatment-experienced patients. Baseline factors, including age, gender, viral load and IL28B GT did not seem to affect the SVR rates. However, the presence of cirrhosis negatively influenced the treatment outcomes, as SVR12 was 63% in patients with cirrhosis (n = 20/32) compared with 96% in those without (n = 105/109).[35] Virological relapse occurred in nine treatment-naïve (9%) and seven treatment-experienced patients (14%), and in these 16 patients, 11 had cirrhosis and nine had emergence of NS5A-Y93H variants.[35] In addition, a real-life French cohort of 409 GT1 infected patients reported that a combination of DCV plus SOF, with or without RBV, was associated with a high rate of SVR4. The addition of RBV increased the SVR rate in cirrhotic or experienced patients (to near 100%) without additive benefit of the extension of the treatment from 12 to 24 weeks.[36]
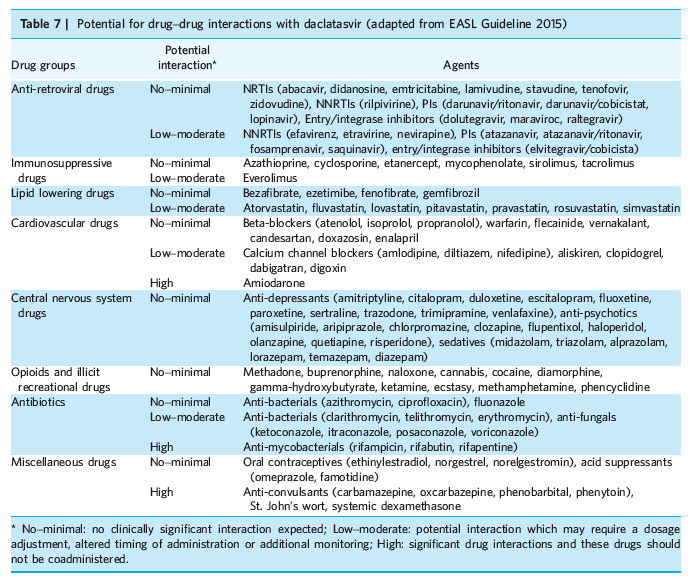
Drug-drug interactions (DDI) between DCV other medications appear to be minor. Approximately 90% of DCV is eliminated in faeces (half as unchanged drug) and less than 10% is excreted in the urine (primarily as unchanged drug).[1] DCV is metabolised by hepatic CYP3A4, but without significant clinical evidence of CYP3A4 inhibition or induction.[12] DCV is a moderate inhibitor of P-glycoprotein (P-gp) and OATP1B1, and thus there is a potential for mild DDI with P-gp and OATP1B1 substrates.[12] Coadministration of DCV with drugs that strongly induce CYP3A4 and P-gp can reduce DCV exposure, and therefore such drugs should be avoided. This includes anti-convulsants (carbamazepine, phenytoin, oxcarbazepine, phenobarbital), anti-mycobacterials (rifampicin, rifabutin, rifapentine), systemic dexamethasone and St John's wort.[13, 53] The dose of DCV should also be reduced to 30 mg with clarithromycin, telithromycin, erythromycin, ketoconazole, itraconazole, posaconazole and voriconazole. No dose adjustment is required when coadministrating DCV with acid-reducing agents (famotidine, omeprazole), escitalopram and oral contraceptives.[13, 53, 54] However, due to DCV inhibiting some transport proteins, monitoring is required with dabigatran and digoxin and other P-gp substrates.[53] The potential for DDI between DCV and commonly used medications are summarised in Table 7.[13, 53] In addition, exposures of DCV appeared to be unaffected when coadministered with SOF, and no dose adjustment is required for either compound in combination.[55]
------------------------------------------
Review article: the efficacy and safety of daclatasvir in the treatment of chronic hepatitis C virus infection
Summary
Background
The treatment of hepatitis C virus (HCV) has evolved dramatically after the introduction of direct acting anti-virals. NS5A protein plays an important role in HCV replication and is an attractive target for drug development.
Aim
To review clinical studies on the efficacy and safety of direct-acting anti-virals regimens containing daclastavir, an NS5A inhibitor, in the treatment of chronic hepatitis C.
Methods
A Medline search was undertaken to identify relevant literature using search terms including 'daclatasvir', 'HCV treatment' and 'NS5A inhibitors'.
Furthermore, we scanned abstracts presented at the recent international meetings in liver disease, viral hepatitis and infectious disease, as well as the reference lists of the review articles to identify publications not retrieved by electronic searches.
Results
Daclatasvir is the first-in-class HCV NS5A inhibitor that has been demonstrated in Phase I-III trials to have a potent anti-viral effect and clinical efficacy across multiple HCV genotypes (GT). Daclastavir is generally safe and well tolerated, with a low barrier to resistance and low potential for drug-drug interaction. When Daclastavir is added to PEG-IFN/RBV platform, sustained virological response (SVR) rates are increased significantly compared with PEG-IFN/RBV alone. The all-oral combination of Daclastavir/asunaprevir (ASV; protease inhibitor) has high SVR rates against GT1b, but less activity against GT1a. Dual combination of Daclastavir/Sofosbuvir (SOF; nucleotide polymerase inhibitor) and triple combination of Daclastavir/ASV/beclabuvir (BCV; non-nucleoside polymerase inhibitor) have demonstrated >90% SVR rates in both treatment naïve and treatment-experienced patients with GT1. Furthermore, Daclastavir/SOF combination has also demonstrated up to 90% SVR rates in patients with GT3, and in those with human immunodeficiency virus coinfection, cirrhosis and post-transplant HCV recurrence with any GT. Daclastavir/ASV/BCV has primarily demonstrated near 100% SVR rates in patients with GT4.
Conclusion
Daclastavir-containing regimens, with or without PEG-IFN, have shown promising results in clinical trials, and present an excellent treatment option for those with chronic HCV and for multiple genotypes.
Introduction
Worldwide, chronic hepatitis C virus (HCV) infection is a leading cause of chronic liver disease and hepatocellular carcinoma. Treatment with pegylated interferon (PEG-IFN) and ribavirin (RBV) had been the standard of care for HCV patients for a decade, until the development of several direct-acting antivirals (DAA), that are in three main classes; NS3/4A inhibitors [e.g. boceprevir (BOC), telaprevir (TPV), simeprevir, asunaprevir (ASV), and paritaprevir boosted by ritonavir], NS5A inhibitors [e.g. daclatasvir (DCV), ledipasvir, and ombitasvir], and NS5B nucleotide [sofosbuvir (SOF)] and non-nucleoside (e.g. dasabuvir) polymerase inhibitors. These DAAs have been approved variably in various parts of the World as either along with PEG-IFN and RBV, or with RBV, or in combination as multiple all oral DAAs with or without RBV, while new DAAs particularly of a pangenotypic nature are being developed. DCV have been approved for the treatment of HCV genotype (GT) 1 as part of three-drug combination with PEG-IFN and RBV, and along with ASV in certain regions of the World. In addition, the recent European Association of the Study of the Liver (EASL) recommendations have suggested the use of DCV-containing regimens as an option for all HCV GTs in various groups patients, including treatment-naïve, treatment-experienced, compensated and decompensated cirrhosis, chronic kidney disease, post-liver transplantation (LT) and human immunodeficiency virus (HIV) coinfection.[1]
The excellent tolerability and high sustained virological response (SVR) rates with all oral therapy for HCV infection in the clinic could signal the end of the need for IFN as an integral component of the standard of care. This transition would reduce the burden of treatment in all HCV patients and allow more individuals to be treated including those who are intolerant of interferon or are unresponsive, and further have contraindications to IFN-based therapy.
This review will walk you through the development of the first-in-class NS5A inhibitor, DCV, from the in vitro and clinical phase study stages to an official approval. In addition, the safety and efficacy of DCV in various treatment regimens and patient populations will also be discussed.
Mechanism of actions
The HCV genome encodes for 10 polyproteins: three structural proteins are found off the N-terminus while seven nonstructural (NS) proteins are off the C-terminus.[2] Three of the non-structural proteins, that include NS3/4A, NS5A, and NS5B, are important for HCV replication and therefore make them attractive targets for suppression.[2, 3] NS3/4A is a serine protease critical for viral replication. Inhibitors of NS3/4A, such as TPV, BOC, simeprevir, paritaprevir, grazoprevir and ASV, are characterised as having a high potency but a low barrier to resistance and may or may not target multiple HCV GTs.[2, 3] NS5B is an HCV RNA-dependent RNA polymerase that is also vital for HCV replication. As the catalytic site of NS5B is highly conserved across all HCV GTs, nucleotide inhibitors such as SOF have been associated with pan-genotypic activity and possess a high potency and high barrier to resistance.[2, 3] Non-nucleoside NS5B polymerase inhibitors, such as dasabuvir and beclabuvir (BCV, formerly BMS-791325), target allosteric sites on NS5B and display a low barrier to resistance, mild potency and limited effectiveness across all HCV GTs.[2, 3]
NS5A is a 447 amino acid, zinc-binding phosphoprotein that plays an important but currently unclear role in HCV replication. Unlike the former two proteins, it has no known enzymatic functions. NS5A is comprised of three distinct structural domains as well as an amphipathic α helix at its N terminus that functions in membrane localisation.[1, 4, 5] Domain I is essential for viral RNA replication and has been crystallised as a homodimer. Domains II and III are less well characterised. Domain II is involved with binding to cyclophilin A and has been postulated to play a role in antagonising the innate immune response to HCV. Domain III seems to be important for the assembly of infectious viral particles. As NS5A is involved in multiple steps in HCV replication, inhibitors of NS5A are believed to have potent anti-viral activity, however, they appear to have relatively low barrier to resistance.[1, 4, 5] DCV is the first-in-class HCV NS5A inhibitor which has shown significant promise in clinical trials.[2] Subsequently, other NS5A inhibitors, such as Ledipasvir and ombitasvir has been evaluated and approved for use while GS-5816 and elbasvir, are under clinical investigation.
Pharmacologic properties
The identification of DCV as a potent inhibitor of NS5A began with the goal of finding a compound functionally distinct from NS3/4A and NS5B inhibitors.
Screening of over 1 million compounds from the Bristol-Myers Squibb (BMS) proprietary collection yielded the iminothiazolidinone BMS-858, a pre-cursor to DCV, as a weak but specific inhibitor of HCV replication.[6, 7] This compound served as the foundation for a series of chemical manipulations that improved anti-viral potency, expanded the GTs impacted and improved pharmacokinetic properties and oral bioavailability.[6, 7]After defining symmetry as an important contributor to anti-viral activity, DCV (formerly labelled BMS-790052) was identified as a development candidate for advancement into clinical trials.[7] Further investigations into the activity of DCV demonstrated that the target for inhibition was the first 100 amino acids on the NS5A protein.[2, 7]
The mechanism by which DCV inhibits viral replication remains unclear. A speculation has been that on binding to NS5A, DCV causes small structural distortions that directly or allosterically inhibit the protein's function.[2, 7] Alternatively, DCV could inhibit viral replication by inhibiting two or more activities of NS5A that interact synergistically.[2, 7] In vitro studies suggested that DCV could interfere with protein-protein interactions at the stage of membranous web biogenesis, which can explain potent inhibition of replication-complex formation, resistance, effects on lipid droplet distribution and virion release.[2, 7-9] Accordingly, a mathematical modelling of viral kinetics during therapy demonstrated an initial, rapid viral decline followed by a slower fall in HCV-RNA, confirming an inhibiting effect of DCV not only on viral replication but also on viral assembly and secretion.[10]
Preclinical and phase I studies
In vitro studies showed that it is highly potent with picomolar half-maximum effective concentrations (EC50) in replicons ranging from 9 to 146 pmol\L, and expressing a broad range of anti-viral activity across HCV GTs with higher activity against GTs 1 (1b higher than 1a), 4 and 5 GT1b and lower activity against GTs 2 and 3.[7] In addition, DCV displayed additive-to-synergistic effects with interferon, RBV and inhibitors of NS3/4A protease and NS5B polymerase, which indicates the potential for DCV as a candidate for combination therapy with other HCV therapeutic agents.[7]
Phase I studies demonstrated that DCV was well-absorbed, safe over a range of 1-200 mg and could be administered as a once daily regimen.[7, 11] DCV alone has revealed very encouraging anti-viral properties in vivo as HCV-infected individuals experienced an approximate 3 log10 decline in serum HCV-RNA after the initial dose (Table 1).[7, 11] However, only 13% of patients reported undetectable levels at the end of treatment.[7, 11] Similar rates of adverse events were reported among placebo and treatment groups with headache reported most frequently.[7, 11] In addition, there was no food effect and no dose adjustment is required for patients with any degree of hepatic and renal impairment.[7, 12, 13]
Phase II studies
IFN-based regimens (Table 2)
Several phase II studies of DCV have been conducted both in the IFN-based and in the all-oral regimens. Overall results from IFN-based treatment studies demonstrated a moderate to high SVR rates (60-100%) in naïve patients with HCV GT1 when DCV at a dosage of >10 mg was added to the regimen (60 mg of DCV was noted to be an optimal dose).[14-17] Addition of DCV to the PEG-IFN/RBV platform was associated with higher SVR rates compared with PEG-IFN/RBV alone, although the differences were not very impressive and no P-values were provided in some studies.[14-16] The largest Phase II study was the COMMAND-1 study which included treatment-naïve patients with GT1 (n = 365) and GT4 (n = 30) randomised to treatment with DCV 20 mg, 60 mg or placebo (2:2:1), along with PEG-IFN and RBV for 24 weeks.[16] Overall, GT1 patients achieved SVR24 rates of 62%, 60% and 38% for the 20 mg, 60 mg and placebo groups respectively. GT1a patients attained SVR24 at rates of 57%, 55% and 36%, whereas GT1b patients attained SVR24 at rates of 76%, 77% and 44% for the 20 mg, 60 mg and placebo groups respectively.[16] Patients with GT4 in the 20 mg, 60 mg and placebo groups achieved SVR24 rates of 67%, 100% and 50% respectively.[16] Notably, an important factor influencing treatment response that was alluded to during pre-clinical studies and has been confirmed in Phase II studies is the difference in SVR between GT1a (55-57%) and GT1b (76-77%) patients treated with DCV.[14-18] This may be explained largely by a lower genetic barrier to resistance for DCV in GT1a as noted by higher on-treatment virological failure due to emergence of DCV-associated resistant variants (GT1a: 21% vs. GT1b: 8%).[16, 18] These rates of SVR in GT 1 patients with DCV, PEG-IFN and RBV, are similar to the efficacy rates reported by real-life data on the first-generation protease inhibitors, TPV and BOC, which achieved 68% and 56% SVR rates, respectively, in cohorts of patients enriched with factors associated with treatment failure, such as advanced fibrosis, older age and concomitant comorbidities.[18-20] However, DCV clearly demonstrates an advantage over TPV and BOC in terms of safety and tolerability profiles.[18-20] Higher SVR rates have been demonstrated when ASV is incorporated to the DCV/PEG-IFN/RBV regimen.[21, 22]
The triple combination of DCV/PEG-IFN/RBV appears to perform less well in patients with GT2 or GT3.[23] The Phase IIb GT2/3 Study included 151 patients randomised to DCV plus PEG-IFN/RBV for 12 or 16 weeks or placebo plus PEG-IFN/RBV for 24 weeks. SVR24 was achieved by 83%, 83% and 63% of GT2 patients in the 12 weeks, 16 weeks and placebo groups respectively.[23] Patients with GT3 achieved SVR24 at rates of 69%, 67% and 59% in the 12 week, 16 week and placebo cohorts respectively.[23] [Corrections added on 29 June 2015 after first online publication : In this paragraph, the rates for the GT2 patients in the 12 weeks and all the rates for GT3 were previously wrong and have now been corrected.]
All-oral regimens (Table 3)
Several Phase II studies have been conducted evaluating the efficacy of HCV anti-viral treatment without IFN that included DCV and one or two additional DAAs. A combination of DCV and ASV has shown mixed results in HCV GT1 patients who failed treatment previously as this combination seems to work very well in Japanese patients with GT1b, but not in American patients with GT1a.[21, 22, 24, 25] In Suyuki et al., SVR was achieved by 90.5% and 63.6% of null responders (n = 21) and IFN ineligible/intolerant (n = 22) patients respectively.[25] Although in Lok et al. study, the SVR rates associated with DCV/ASV combination were less than 40%.[21]
Daclatasvir and SOF, with and without RBV, as combination therapy has demonstrated impressive efficacy in a large Phase II study by Sulkowski et al.[26] This study initially randomly assigned 44 previously untreated patients with GT1 and 44 patients with GT2/3 to DCV plus SOF, with or without RBV, for 24 weeks. Subsequently, the study was expanded to include 123 additional patients with GT1 to be treated with DCV plus SOF, with or without RBV, for 12 weeks (82 naïve patients) or 24 weeks (41 patients who had previous failed TPV or BOC plus PEG-IFN/RBV).[26] All naïve GT1 patients in the 24-week treatment cohorts achieved SVR24 while GT1 patients in the 12 week groups achieved SVR24 at rates of 93-95%.[26] The SVR rates were 88-100% in naïve patients with GT2/3 and were 95-100% in patients with GT1 who previously failed protease inhibitors.[26] Notably, SVR rates were not related to IL28B status, GT subtype or the administration of RBV.[26]
To improve response rates, DCV, ASV and BCV (formerly BMS-791325 and a potent NS5B non-nucleoside inhibitor) have been evaluated in a Phase II study.[27, 28] The overall SVR rates of this triple combination for naïve and treatment-experienced GT1 patients were high (88-100%), particularly when BCV was administered at dose 150 mg b.d.[27, 28] There was no apparent difference in SVR rates between those treated for 12 or 24 weeks.[27, 28] Subsequently, a 12-week course of this triple combination has been evaluated in 21 naïve patients with GT4 and yielded SVR rates of 100% with BCV dose of either 75 or 150 mg b.d.[29]
Phase III and Phase IV studies
IFN-based regimens (Table 4)
The HALLMARK-QUAD study has evaluated the efficacy of a 24-week course of DCV/ASV/PEG-IFN/RBV in treatment-experienced patients with GT1 (n = 354) and GT4 (n = 44).[30] Among GT1 patients, 50% were GT1a, 91% were IL28B non-CC GTs, and 66% had previous null response. The overall SVR12 rates were excellent (87% for GT1a, 99% for GT1b and 100% for GT4).[30] In COMMAND-4 study, a response-guided therapy with DCV/PEG-IFN/RBV in treatment-naïve GT4 patients (n = 82) showed SVR12 of 82%, compare to 43% in those who were randomised to receive 48-week PEG-IFN/RBV.[31] In the response-guided arm, DCV-treated patients with undetectable HCV-RNA at weeks 4 and 12 (eRVR) received 24 weeks of DCV/P/R and those without an eRVR received an additional 24 weeks of P/R (79% of patients in this study required 24 weeks of treatment).[31] The results of more Phase III studies evaluating DCV plus PEG-IFN/RBV in patients with GT1 (COMMAND-3) and DCV plus lamda-IFN/RBV in patients with GT1-3 (PRINCIPAL and STRUCTURE), as well as in patients with haemophilia and HIV coinfection, are awaited.
All-oral regimens (Table 5)
The first published Phase III study was the HALLMARK-NIPPON study evaluating a combination of DCV plus ASV in 222 Japanese patients with HCV GT1b who either failed to respond (n = 87), were ineligible (n = 100) or were intolerant (n = 35) to previous PEG-IFN/RBV therapy.[32] All patients were administered DCV 60 mg daily plus ASV 100 mg b.d. (lower dose than that used in previous Phase II trials) for 24 weeks. Overall, 85.1% (n = 189/222) achieved SVR12, with 84.7% (n = 188/222) achieving SVR24; 80.5% of prior non-responders vs. 87.4% of IFN-intolerant/ineligible patients achieved SVR24.[32] There was no significant difference between IL-28B CC and non-CC patients. Among 22 patients with Child's A cirrhosis, 90.9% (n = 20) of patients achieved SVR24. Fifteen patients discontinued therapy due to lack of efficacy, 11 due to adverse events and 2 for unspecified reasons.[32]Another Phase III study of Japanese patients with GT1b using similar regimen noted slightly better results with SVR12 of 89% in naïve patients and 96% in relapsers.[33] A shorter duration of treatment with DCV 60 mg daily plus ASV 100 mg b.d. for 12 weeks was evaluated in the large multinational Phase III study (HALLMARK-DUAL) which included 307 treatment-naive patients, 205 non-responders and 235 ineligible, intolerant or ineligible and intolerant patients with HCV GT1b from 18 countries.[34] SVR12 was achieved in 90% of treatment-naive, 82% of non-responder, and 82% of ineligible and/or intolerant cohorts.[34] Similar to previous trials, majority of patients (68-83%) achieved undetectable HCV-RNA after week 4 of treatment. Again, there were no differences in SVR rates based on sex, age, race, body-mass index, IL28B GT or the presence of cirrhosis (SVR12 rates were 84% in patients with cirrhosis and 85% in those without).[34]
Due to the impressive results of DCV/SOF combination for GT1 and GT3 in Phase II study,[26] this dual combination was again evaluated without RBV in a Phase III study (ALLY-3) which included 101 treatment-naïve and 51 treatment-experienced patients with HCV GT3.[35] All patients received a 12-week regimen of once daily DCV 60 mg plus SOF 400 mg, which resulted in SVR12 in 90% of treatment-naïve and 86% of treatment-experienced patients. Baseline factors, including age, gender, viral load and IL28B GT did not seem to affect the SVR rates. However, the presence of cirrhosis negatively influenced the treatment outcomes, as SVR12 was 63% in patients with cirrhosis (n = 20/32) compared with 96% in those without (n = 105/109).[35] Virological relapse occurred in nine treatment-naïve (9%) and seven treatment-experienced patients (14%), and in these 16 patients, 11 had cirrhosis and nine had emergence of NS5A-Y93H variants.[35] In addition, a real-life French cohort of 409 GT1 infected patients reported that a combination of DCV plus SOF, with or without RBV, was associated with a high rate of SVR4. The addition of RBV increased the SVR rate in cirrhotic or experienced patients (to near 100%) without additive benefit of the extension of the treatment from 12 to 24 weeks.[36]
The UNITY-1 Phase III study evaluated triple combination of DCV, ASV and BCV in 312 treatment-naïve and 103 treatment-experienced patients with HCV GT1 patients.[37] All patients received a 12-week course of twice-daily, fixed-dose combination tablet of DCV 30 mg, ASV 200 mg and BCV 75 mg. In treatment-naïve cohort, SVR12 rates were 90% in GT1a and 96% in GT1b, whereas in treatment-experienced cohort, SVR12 rates were 85% in GT1a and 100% in GT1b.[37] This triple combination was well-tolerate with low rates of serious adverse events (2%) and discontinuation (0.7%).[37]
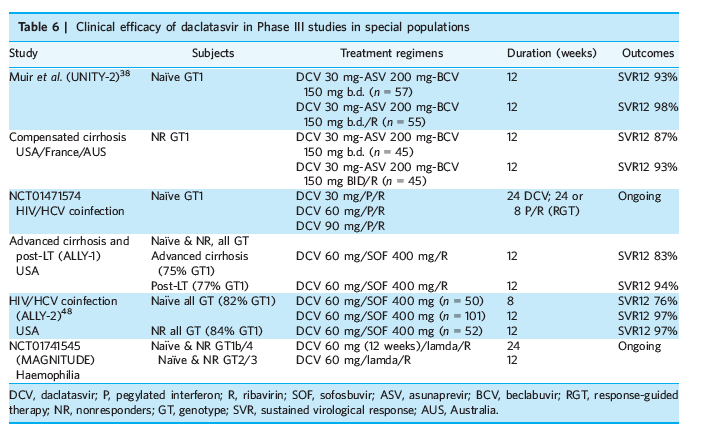
For special populations (Table 6)
Due to its favourable safety and DDI profile, DCV has potential for use in patients with decompensated cirrhosis, renal impairment, post-LT and HIV coinfection. Several Phase III studies are being conducted to assess the safety and efficacy of DCV-containing regimens for HCV patients with cirrhosis (UNITY-2 and ALLY-1), HIV coinfection (ALLY-2 and NCT01471574), LT (ALLY-1) and haemophilia (MAGNITUDE). Results of the UNITY-2 trial revealed high SVR12 rates after 12 weeks of treatment with DCV, ASV and BCV, with or without RBV, in 202 GT1 patients with compensated cirrhosis (Child-Pugh A, platelets >50 000/mm3, INR <1.7 and albumin >3.5 g/dL).[38] SVR12 rates were 93-98% among naïve patients (n = 112), and 87-90% in treatment experienced patients (n = 90). Addition of RBV appeared to slightly decrease frequency of relapse in patients with GT1a. With RBV, in treatment-naïve cohort, SVR12 rates were 97% in GT1a and 100% in GT1b, whereas in treatment-experienced cohort, SVR12 rates were 91% in GT1a and 100% in GT1b. Without RBV, in treatment-naïve cohort, SVR12 rates were 90% in GT1a and 100% in GT1b, whereas in treatment-experienced cohort, SVR12 rates were 86% in GT1a and 90% in GT1b.[38]
The Phase III ALLY-1 study evaluated a 12-week course of DCV 60 mg daily, SOF 400 mg daily and RBV (initially 600 mg/day, adjusted to 1000 mg/day based on haemoglobin levels) in HCV GT1-6 patients (about 70% were GT1) with advanced cirrhosis (n = 60) or post-LT HCV recurrence (n = 53).[39] In advanced cirrhosis cohort, SVR12 was achieved in 92%, 94% and 56% of patients with Child-Pugh class A (n = 12), B (n = 32), and C (n = 16) respectively.[39] Majority of patients improved their MELD scores following treatment, though some patients continued to show further increases in MELD score. In post-LT cohort, SVR12 was achieved in 92% of patients without the need of dose modification of immunosuppressive agents. Among patients with GT3 (n = 17), SVR12 rate was 83% in advanced cirrhosis and 91% in post-LT cohort.[39] An interim analysis of Phase II (SATURN) study, evaluating a 24-week course of DCV 60 mg daily, simeprevir 150 mg daily and RBV 1000-1200 mg/day for LT recipients with recurrent HCV GT1b, demonstrated the best on-treatment response and SVR4 (90-93%).[40]
In addition, several compassionate-use programs in Europe and USA have demonstrated high efficacy and favourable safety profile of DCV-containing regimens in LT recipients with severe recurrent HCV, including fibrosing cholestatic hepatitis.[41-47] The treatment regimens were well-tolerated and calcineurin inhibitors trough levels were within the targeted range throughout and after treatment.[45, 46] In the large French prospective ANR CO23 CUPILT study, 116 LT recipients with severe HCV recurrence treated with a 24-week course of DCV plus SOF, with or without RBV, achieved SVR12 rates of 97% and 96% respectively.[43] Twenty-three patients with post-LT fibrosing cholestatic HCV GT1-4 received DAA-based therapy for 24 weeks and SVR12 was attained in 88% of patients taking SOF plus RBV, with or without PEG-IFN, (n = 8) and 100% taking SOF plus DCV, with or without RBV (n = 15).[44] Overall, 87% of patients achieved complete clinical response, defined as being still alive without re-LT, normalised bilirubin, and no ascites or hepatic encephalopathy. Half the participants experienced serious adverse events, but none were attributed to SOF or DCV.[44]
The clinical efficacy of DCV plus SOF in HIV/HCV coinfected patients has been evaluated in Phase III study (ALLY-2), which included 151 treatment-naïve patients and 52 treatment-experienced patients with HIV/HCV coinfections (83% GT 1, 9% GTs 2, 6% GT 3, and 2% GT 4).[48] The standard dose of DCV was 60 mg/day with dose-adjusted for concomitant anti-retroviral therapy (30 mg with ritonavir-boosted protease inhibitors, 90 mg with NNRTIs except rilpivirine). Overall, 97% of patients achieved SVR12 after 12 weeks treatment, compared with 76% in those who were treated for 8 weeks. Factors associated with relapse included high baseline HCV-RNA (>2 000 000 IU/mL), presence of cirrhosis and coadministration with darunavir-boosted anti-retroviral regimen (with DCV 30 mg/day).[48] In addition, an interim analysis of a French multicenter cohort has confirmed the safety and efficacy of DCV plus SOF, with or without RBV, for 12-24 weeks in 733 HIV/HCV coinfected patients, including those with advanced liver disease.[49]
Safety and tolerability
Side effects and adverse events
Daclatasvir has been reported to be safe and well-tolerated in combination with other anti-virals and DAAs such as PEG-IFN/RBV, ASV, BCV and SOF.[2, 14-16, 24-28, 34] Thus, serious adverse events directly related to DCV have not been reported so far. Fatigue (43-45%) and headache (33-41%) are the most common side effects when DCV is administered with PEG-IFN/RBV, with incidence rates similar to that of PEG-IFN/RBV alone.[14-16, 23] Diarrhoea appears to be the more common complaint when ASV is added to DCV.[24, 25, 28, 34] In Phase III study of all-oral DCV/ASV combination, headache, fatigue, diarrhoea, nausea and asthenia occurred in 24-25%, 21-22%, 12-22%, 11-12% and 2-11% of patients at the end of treatment respectively.[32-34]
It should be noted that a combination of DCV plus ASV has been associated with self-limiting elevations of serum alanine aminotransferase (ALT) in about 5-29% of patients in Phase II and III studies (with higher rates at dose >200 mg b.d. or when coadministered with PEG-IFN/RBV).[21, 22, 24, 25, 30, 32-34] This phenomenon is rarely associated with increased serum bilirubin or the need of treatment discontinuation, and it appears to be attributable to ASV rather than DCV. In a Phase 2b study of ASV, treatment-naive patients with GT1 (n = 213) or GT4 (n = 25) were randomly assigned (3:1) to ASV 200 mg or placebo twice daily plus PEG-IFN/RBV.[50] Grade 3-4 ALT elevation was encountered in 10% of patients in the ASV arm and in 2% in the placebo arm.[50] Further, in the D-LITE study that evaluated PEG-IFN lamda/RBV combined with either DCV 60 mg daily or ASV 200 mg twice daily for 119 naïve HCV GT1 patients, grade 3-4 ALT elevation were more frequently recorded in the ASV arm (6-9%) than in the DCV arm (0-5%).[51]
Notably, serum ALT elevation of >5 times limit of normal was observed in 5% of patients in the Phase III study (UNITY-1) of a triple combination of DCV/ASV/BCV with 2/415 patients discontinuing therapy.[37]
In an open-label, Phase III study of DCV plus ASV conducted in Japan, serum ALT elevations occurred in 16% and were above five times the upper limit of normal in 7% of patients.[32] In two patients, serum ALT elevations were accompanied by fever and eosinophilia, suggesting immunoallergic hepatitis.[52] In all except one, the ALT elevations resolved promptly upon discontinuation of the therapy and patients achieved a SVR. In the one case, hypersensitivity reaction accompanied by liver injury and jaundice persisted for a week after stopping the anti-viral agents and this led to the use of corticosteroids.[52] A liver biopsy performed on day 36 revealed inflammatory infiltrates of eosinophils, lymphocytes and plasma cells in the hepatic lobules and portal areas, with focal lobular necrosis, interface hepatitis and bridging fibrosis. The overall clinical syndrome was typical of drug-hypersensitivity syndrome.[52] Whether DCV or ASV was responsible for the hypersensitivity reaction is unclear. Interestingly, this syndrome as well as ALT elevations and fever appear to be more frequent in Japanese[32] than USA or European studies of these agents,[22, 34] suggesting genetic background on susceptibility of the reaction.
Drug-drug interactions
Drug-drug interactions (DDI) between DCV other medications appear to be minor. Approximately 90% of DCV is eliminated in faeces (half as unchanged drug) and less than 10% is excreted in the urine (primarily as unchanged drug).[1] DCV is metabolised by hepatic CYP3A4, but without significant clinical evidence of CYP3A4 inhibition or induction.[12] DCV is a moderate inhibitor of P-glycoprotein (P-gp) and OATP1B1, and thus there is a potential for mild DDI with P-gp and OATP1B1 substrates.[12] Coadministration of DCV with drugs that strongly induce CYP3A4 and P-gp can reduce DCV exposure, and therefore such drugs should be avoided. This includes anti-convulsants (carbamazepine, phenytoin, oxcarbazepine, phenobarbital), anti-mycobacterials (rifampicin, rifabutin, rifapentine), systemic dexamethasone and St John's wort.[13, 53] The dose of DCV should also be reduced to 30 mg with clarithromycin, telithromycin, erythromycin, ketoconazole, itraconazole, posaconazole and voriconazole. No dose adjustment is required when coadministrating DCV with acid-reducing agents (famotidine, omeprazole), escitalopram and oral contraceptives.[13, 53, 54]
However, due to DCV inhibiting some transport proteins, monitoring is required with dabigatran and digoxin and other P-gp substrates.[53] The potential for DDI between DCV and commonly used medications are summarised in Table 7.[13, 53] In addition, exposures of DCV appeared to be unaffected when coadministered with SOF, and no dose adjustment is required for either compound in combination.[55]
Although there is a potential for modest DDI with calcineurin inhibitors and some anti-retroviral agents, the clinical safety and favourable DDI profiles of DCV in LT recipients and patients with HIV coinfection have been demonstrated in Phase III studies (ALLY-1 and ALLY-2).[39, 48] As a CYP3A4 substrate, the plasma level and total dose exposure to DCV is altered when coadministered with some anti-retroviral agents that are strong inducers or inhibitors of CYP3A4. With efavirenz (an enzyme inducer), the dose of DCV is recommended to be increased to 90 mg once daily.[56] With atazanavir/ritonavir and cobicistat containing anti-retroviral regimens (enzyme inhibitors), the dose of DCV should be reduced to 30 mg once daily.[56]In the ALLY-2 study in HIV-coinfected patients receiving DCV plus SOF, patients on a darunavir-based regimen who had DCV dose reduced to 30 mg (based on the original atazanavir/ritonavir study data) had a reduced rate of SVR12 (93%), particularly in the 8-week treatment arm (67%).[48] There are data to suggest that no dose adjustment of DCV is necessary with either darunavir/ritonavir or lopinavir/ritonavir.[53]
Resistance
The overall genetic barrier to resistance of DCV is low-to-moderate as substitutions at one or two base pairs of NS5A can be sufficient to confer HCV resistance to DCV. In vitro and in vivo resistance analysis of DCV has revealed key viral factors that could impact treatment outcomes and the major resistance variants of each GT are summarised in Table 8.[4, 5, 57] The rank order of resistance barriers to DCV is 1b > 4a > 5a > 6a > 1a > 2a(JFH) > 3a > 2a(M31).[57] In patients with GT1, L31 and Y93 substitutions appear to be the most important and commonly observed resistance-associated polymorphisms in both GT1a and GT1b, but a greater number of mutations in GT1a than in GT1b are required to confer resistance.[2, 4, 5, 57, 58]
Fortunately, DCV-resistant variants have been shown to remain sensitive to IFN and other DAAs. Thus, the emergence of DCV-related mutations can potentially be preventable by anti-viral pressure of additional DAAs.[2, 59] The presence of DCV resistance-associated polymorphisms at baseline may increase, but do not invariably increase the risk of virological failure.[2, 59, 60] In combination treatment with two DAAs, patients with virological failure were typically found to harbour HCV with resistance to both agents by the end of treatment.[2, 59] Notably, a baseline resistance analysis of >2000 GT1 patients in Phase II and III studies of SOF plus ledipasvir revealed that baseline NS5A resistance-associated variants are quite frequent and at around 15% in either the USA or European population.[61] However, the combination of NS5A inhibitors plus NS5B nucleotide inhibitors with a high barrier to resistance and a potent anti-viral activity can overcome this problem and lead to a high rate of SVR as demonstrated in the SOF plus ledipasvir combination studies.[62, 63]
One good example of virological response and resistance pattern can be learnt from the studies evaluating DCV plus ASV in HCV patients with GT1b. In the Phase III study in naïve GT1b patients, the presence of NS5A resistance variants (at L31 or Y93) was a negative predictor of SVR12 in multivariate analysis.[34]
Baseline L31 variants were present in 27 (5%) patients, 11 (41%) of whom achieved SVR12, and NS5A-Y93 variants were present in 48 (8%) patients, 18 (38%) of whom achieved SVR12.[34] In the Phase III study of 222 treatment-experienced GT1b patients treated with DCV plus ASV, of the 34 patients with virological failure, 29 had resistance-associated substitutions to both DCV (predominantly L31M/V-Y93H) and ASV (predominantly D168) detected at failure and 22 patients with virological failure had NS5A polymorphisms L31M/V and/or Y93H prior to treatment.[32] Of interest is that 37 patients with L31M/V and/or Y93H at baseline, 11/23 IFN-ineligible/intolerant patients and 4/14 nonresponders achieved SVR.[32] In addition, in vitro study assessing retreatment options for patients with HCV GT1b resistant to DCV plus ASV suggested a number of potential all-oral treatment options with comparable activity against wild-type and DCV/ASV-resistant cell lines.[64] These potential options are combinations of DCV plus SOF, SOF plus ledipasvir, SOF plus simeprevir and DCV, ASV plus BCV.[64]
Drug approval and future perspectives
Through evidence from multiple clinical trials, DCV has been approved by two stringent regulatory authorities to date. In Japan, DCV was approved as a dual combination of DCV and ASV in July 2014, bringing to Japanese patients with HCV the first all-oral, IFN-/RBV-free treatment regimen. Subsequently in August 2014, DCV was approved by the European Commission (EC) and the European Medicines Agency for use as combination therapy with other agents across GTs 1, 2, 3 and 4 for the treatment of HCV. These approvals led to the marketing of DCV (Daklinza; Bristol-Myers Squibb, Inc., Princeton, NJ, USA) in Japan and European countries. In the USA, DCV is not FDA approved and thus the use of it is limited to clinical trials and compassionate use programs.
The new HCV treatment paradigm clearly aims towards all-oral regimens, preferably with pangenotypic activity and not more than 12 weeks treatment duration, and with >95% success rates. DCV has relatively low barrier to resistance so that additional potent DAA is (are) required to achieve this goal. Phase II and III studies have shown that DCV works very well in combination with SOF and currently is one of the best available, once-daily, all-oral regimen, and with multiple genotypic activity (SVR 93-95% for GT1 and 86-90% for GT3).[53] The 2015 EASL Guideline has recommended the use of this combination as an option for various HCV GT and patient populations (Table 9).[53] In Phase III studies, a twice-daily single-pill triple combination of DCV, ASV and BCV, so-called BMS-TRIO, demonstrated excellent SVR rates against GT1b and GT4, but again with slightly decreased SVR rates for GT1a (85-90%). Additional information on the BMS-TRIO, as well as other DCV-containing regimens, are eagerly awaited.
Summary
Daclatasvir is the first-in-class HCV NS5A inhibitor which has shown significant promise in clinical trials. It has good anti-viral potency against multiple HCV GTs, but with low barrier to resistance, which can be overcome by adding PEG-IFN and/or other DAAs to the regimens. DCV is generally safe and well tolerated with mild potential for DDI. When DCV is added to PEG-IFN/RBV, SVR rates are increased (approximately up to 60-100% in GT1,[14-16] 80% in GT2,[23] 70% in GT3,[23] and 82% in GT4[31]), from PEG-IFN/RBV alone. The all-oral combination DCV and ASV has high SVR rates against GT1b (80-95%)[24, 32-34] leading to its approval in Japan, but has less activity against GT1a (23-40% in experienced patients)[21, 22] making it less than an ideal combination for GT1a-predominant areas, such as the US and many countries in Europe. Dual combination of DCV/SOF and triple combination and DCV/ASV/BCV have demonstrated >90% SVR rates in patients with GT1, both naïve and treatment-experienced. Furthermore, DCV/SOF combination has also demonstrated up to 90% SVR rates in patients with GT3, and DCV/ASV/BCV has primarily demonstrated near 100% SVR rates in patients with GT4. High SVR rates in Phase III studies in special populations, such as those with decompensated cirrhosis, HIV coinfection and LT recipients, supports the use of these regimens in patients with a chronic HCV and with multiple GTs.
|
|
| |
| |
|
|
|