| |
Efficacy and Safety of Ombitasvir, Paritaprevir, and Ritonavir in an Open-Label Study of Patients With Genotype 1b Chronic Hepatitis C Virus Infection With and Without Cirrhosis
|
| |
| |
Download the PDF here
The PEARL-I study is an ongoing phase 2b, open-label, combination treatment study being conducted at 47 sites in France, Hungary, Italy, Poland, Puerto Rico, Romania, Spain, Turkey, and the United States.
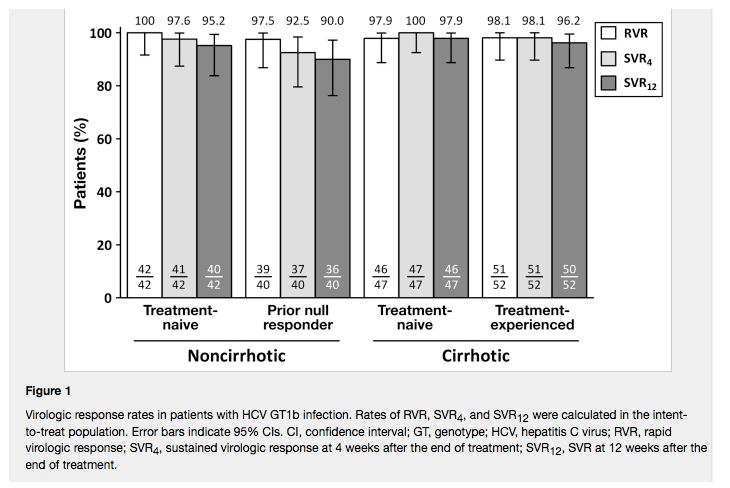
In treatment-naive and null responder patients without cirrhosis, rates of SVR12 were 95.2% and 90.0%, respectively. In treatment-naive and treatment-experienced patients with cirrhosis, rates of SVR12 were 97.9% and 96.2%, respectively
"In a meta-analysis of 992 GT1b-infected patients treated for 12 or 24 weeks, 98.3% of 691 treatment-naive and treatment-experienced patients with or without cirrhosis who received ombitasvir, paritaprevir, and ritonavir plus dasabuvir with ribavirin and 99.3% of 301 treatment-naive and treatment-experienced patients without cirrhosis who received ombitasvir, paritaprevir, and ritonavir plus dasabuvir alone achieved SVR 12 weeks after the end of treatment (SVR12).27 Both regimens were well tolerated, although the ribavirin-free regimen was associated with a lower rate of anemia than the ribavirin-containing regimen (6.5% vs 0.2%).28 Based on these studies, this 3-DAA regimen has been approved for the treatment of patients with HCV GT1a and GT1b infections in the United States and Europe."
Gastroenterology Oct 2015
Eric Lawitz,1 Mihaly Makara,2 Ulus Salih Akarca,3 Paul J. Thuluvath,4 Liliana Lucia Preotescu,5 Peter Varunok,6 Rosa Ma. Morillas,7 Coleen Hall,8 Niloufar Mobashery,8 Rebecca Redman,8 Tami Pilot-Matias,8 Regis A. Vilchez,8 and Christophe Hezode9
1Texas Liver Institute, University of Texas Health Science Center, San Antonio, Texas; 2Outpatient Clinic, Saint Laszlo Hospital, Budapest, Hungary; 3Ege University, Faculty of Medicine, Department of Gastroenterology, Izmir, Turkey; 4Mercy Medical Center and University of Maryland School of Medicine, Baltimore, Maryland; 5National Institute of Infectious Diseases Prof. Dr. Matei Bals , Bucharest, Romania; 6Premier Medical Group, New York Medical College, Poughkeepsie, New York; 7Liver Section and CIBERehd, Department of Gastroenterology, Hospital Universitari Germans Trias i Pujol, Badalona, Spain; 8Infectious Diseases Clinical Development, AbbVie Inc, North Chicago, Illinois; and 9Department of Hepatology and Gastroenterology, Hopital Henri Mondor, AP-HP, Universite Paris-Est, INSERM U955, Creteil, France
--------------------------
Background & Aims
Interferon-free treatment options are rapidly evolving for patients with chronic hepatitis C virus (HCV) genotype 1b (GT1b) infection with cirrhosis and for nonresponders to prior pegylated interferon and ribavirin therapy. We performed a phase 2b, open-label trial of the combination of ombitasvir (a NS5A replication complex inhibitor), paritaprevir, and ritonavir (an NS3/4A protease inhibitor)-an interferon- and ribavirin-free regimen-in difficult-to-treat patients, including prior null responders and patients with cirrhosis.
Methods
In an international study, 82 patients without cirrhosis (42 treatment-naive and 40 prior null responders) and 99 with cirrhosis (47 treatment-naive and 52 treatment-experienced with prior relapse or a null or partial response) with chronic HCV GT1b infection received ombitasvir (25 mg), paritaprevir (150 mg), and ritonavir (100 mg) once daily for 12 weeks (without cirrhosis) or 24 weeks (with cirrhosis). The primary efficacy endpoint was sustained virologic response 12 weeks after the end of treatment (SVR12).
Results
In treatment-naive and null responder patients without cirrhosis, rates of SVR12 were 95.2% and 90.0%, respectively. In treatment-naive and treatment-experienced patients with cirrhosis, rates of SVR12 were 97.9% and 96.2%, respectively. No clinically meaningful differences in rates of SVR12 were observed between patients with or without cirrhosis. Virologic relapse occurred in 3 null responders without cirrhosis and 1 with cirrhosis; virologic breakthrough occurred in 1 null responder without cirrhosis. Common adverse events included headache, asthenia, pruritus, and diarrhea. One patient discontinued taking the drugs because of treatment-related adverse events.
Conclusions
An interferon- and ribavirin-free regimen of ombitasvir, paritaprevir, and ritonavir, achieved high rates of SVR12 in patients with HCV GT1b infection with and without cirrhosis. This regimen was well tolerated and was associated with low rates of treatment discontinuation. ClinicalTrials.gov no: NCT01685203.
Chronic hepatitis C virus (HCV) infection affects up to 150 million people worldwide and is a leading cause of cirrhosis and hepatocellular carcinoma.1 Of the 7 major HCV genotypes (GTs) identified globally,2 GT1 is the most common. GT1b is the most prevalent HCV subtype worldwide, particularly in parts of Europe and Asia, whereas GT1a is more prevalent in North America.3 In the era of interferon-based therapy, HCV GT1 infection has been difficult to treat; approximately 60% of patients do not achieve sustained virologic response (SVR) with pegylated interferon and ribavirin therapy.4, 5
Since the approval of the first direct-acting antiviral (DAA) agents in 2011, HCV therapeutic approaches have rapidly evolved. Although the addition of the NS3/4A protease inhibitors boceprevir, telaprevir, or simeprevir to pegylated interferon and ribavirin regimens increased SVR rates in patients with GT1 infection, efficacy rates have remained low in patients with cirrhosis, particularly in prior null responders (eg, an SVR rate of 19.4% with telaprevir). Furthermore, these pegylated interferon and ribavirin-containing regimens have been associated with additional adverse events (AEs), such as severe rash and a high frequency of anemia.6, 7, 8, 9, 10 The addition of the NS5B inhibitor sofosbuvir to pegylated interferon and ribavirin regimens improved SVR rates (89%) but showed lower efficacy in patients with cirrhosis compared with patients without cirrhosis (80% vs 92%) and in patients with GT1b infection compared with GT1a infection (82% vs 92%).11 Additionally, this regimen has not been studied in treatment-experienced patients, and the AE profile of an interferon-based regimen remains unclear.
The recent development of interferon-free DAA regimens has improved both the efficacy and tolerability of antiviral treatment for HCV, although many such regimens continue to include ribavirin.12, 13 In interferon-free all-oral DAA regimens that include ribavirin, the AE profile of ribavirin is becoming clearer. In several phase 3 trials that compared all-oral DAA regimens with and without ribavirin, treatment discontinuation rates due to AEs were similarly low in the ribavirin-containing and ribavirin-free arms.14, 15, 16, 17, 18 Although ribavirin is associated with several characteristic AEs, such as pruritus, asthenia, and insomnia, most of these events were characterized as mild in severity.16, 17 Furthermore, in recent studies, the frequency and severity of ribavirin-associated anemia appear to be much lower than seen in the past, perhaps due to the absence of the bone marrow-suppressant effects of interferon.19However, the development of ribavirin-free regimens would be an important option for patients who are ineligible to receive or intolerant of ribavirin therapy. Clinical trials in patients with GT1 infection suggest that all-oral DAA regimens may obviate the need for ribavirin as a standard component of therapy and thereby eliminate ribavirin-associated AEs.14, 15, 16, 17, 18, 20, 21, 22
Ombitasvir (formerly ABT-267), an inhibitor of the HCV NS5A protein, and paritaprevir (formerly ABT-450), an NS3/4A protease inhibitor identified by AbbVie and Enanta, have potent antiviral activity against multiple HCV GTs, including 1a and 1b.23 Paritaprevir is administered with the pharmacokinetic enhancer ritonavir (r), which inhibits its metabolism, increasing peak, trough, and overall drug exposures and allowing for once-daily dosing.24 In phase 3 trials, a combination of ombitasvir, paritaprevir, and ritonavir plus dasabuvir (a nonnucleoside NS5B polymerase inhibitor) with or without ribavirin was shown to be effective and well tolerated in treatment-naive and -experienced noncirrhotic patients with HCV GT1 infection.17, 25, 26 In a meta-analysis of 992 GT1b-infected patients treated for 12 or 24 weeks, 98.3% of 691 treatment-naive and treatment-experienced patients with or without cirrhosis who received ombitasvir, paritaprevir, and ritonavir plus dasabuvir with ribavirin and 99.3% of 301 treatment-naive and treatment-experienced patients without cirrhosis who received ombitasvir, paritaprevir, and ritonavir plus dasabuvir alone achieved SVR 12 weeks after the end of treatment (SVR12).27 Both regimens were well tolerated, although the ribavirin-free regimen was associated with a lower rate of anemia than the ribavirin-containing regimen (6.5% vs 0.2%).28Based on these studies, this 3-DAA regimen has been approved for the treatment of patients with HCV GT1a and GT1b infections in the United States and Europe.
PEARL-I is an ongoing, randomized, open-label, phase 2b, combination treatment study evaluating the safety and efficacy of an all-oral interferon-free regimen of ombitasvir, paritaprevir, and ritonavir with or without ribavirin in treatment-naive and pegylated interferon and ribavirin treatment-experienced patients with HCV GT1b or GT4 infection. PEARL-I comprises two substudies; substudy 1 enrolled patients without cirrhosis (HCV GT1b and GT4) and substudy 2 enrolled patients with cirrhosis (HCV GT1b). Results from patients with HCV GT1b infection with and without cirrhosis who received an interferon- and ribavirin-free regimen of ombitasvir, paritaprevir, and ritonavir are reported herein.
Methods
Study Design
The PEARL-I study (ClinicalTrials.gov identifier: NCT01685203) is an ongoing phase 2b, open-label, combination treatment study being conducted at 47 sites in France, Hungary, Italy, Poland, Puerto Rico, Romania, Spain, Turkey, and the United States. Screening for the trial began in August 2012; the last patient completed treatment in March 2014. The study was designed as an open-label study to maximize the probability of all patients in the study achieving SVR.
Additionally, an active comparator group that contained pegylated interferon was not included because it could not be effectively blinded. All GT1b-infected patients without cirrhosis were enrolled and completed treatment before enrollment of the patients with cirrhosis to allow for a sequential evaluation of the 2-DAA regimen in these 2 patient populations. The data reported here are from the primary database lock completed after all patients reached post-treatment week 12. The study was approved by all institutional review boards and was conducted in compliance with the Declaration of Helsinki and International Conference on Harmonisation guidelines. Written informed consent was provided by all patients before enrollment. All authors had access to the study data and critically reviewed, revised, and approved the final manuscript.
Patient Population
Eligible patients were 18 to 70 years of age with chronic HCV GT1b infection (≥6 months) and a plasma HCV RNA level >10,000 IU/mL, and were documented to be without cirrhosis or with compensated cirrhosis defined as Child-Pugh score ≤6 at screening by liver biopsy (Metavir score = 4 or Ishak score >4), FibroScan (Echosens, Paris, France) score ≥14.6 kPa within 6 months of screening or during the screening period, or FibroTest. Subjects with a non-qualifying FibroScan result were enrolled only if they had a qualifying liver biopsy performed during the screening period. Patients without cirrhosis were eligible if they were treatment-naive or had a prior null response to pegylated interferon and ribavirin treatment. Patients with cirrhosis were eligible if they were treatment-naive or if prior pegylated interferon and ribavirin treatment had failed (null/partial response or relapse). Exclusion criteria included co-infection with hepatitis B virus or HIV, liver disease not due to chronic HCV infection, current or past clinical evidence of cirrhosis (in substudy 1), or a Child-Pugh B or C classification or clinical history of liver decompensation such as ascites (noted on physical exam), variceal bleeding, or hepatic encephalopathy (in substudy 2). Detailed eligibility criteria, including definitions of null and partial responses and relapse, are provided in the Supplemental Appendix.
Treatment
All GT1b-infected patients received an oral interferon- and ribavirin-free regimen of ombitasvir, paritaprevir, and ritonavir 25 mg/150 mg/100 mg once daily. The duration of treatment was 12 weeks in patients without cirrhosis and 24 weeks in patients with cirrhosis. The 24-week treatment duration in patients with cirrhosis (substudy 2) was based on initial efficacy and safety data from the prior null responder patients without cirrhosis (substudy 1) and preliminary results from a phase 3 study (TURQUOISE-II) that demonstrated that a 24-week treatment duration with ombitasvir, paritaprevir, and ritonavir plus dasabuvir and ribavirin in HCV GT1-infected patients with cirrhosis led to higher SVR rates than 12 weeks of treatment, particularly in prior null responders.13 After completion or early discontinuation of treatment, patients were followed for 48 weeks to monitor HCV RNA levels, the emergence and persistence of resistant viral variants, and serious AEs.
Efficacy Endpoints and Assessments
Plasma samples were collected at screening and each study visit, and HCV RNA levels were determined by real-time polymerase chain reaction using the COBAS TaqMan® HCV test version 2.0 (Roche Molecular Diagnostics, Indianapolis, IN), which has a lower limit of quantitation of 25 IU/mL and a lower limit of detection of 15 IU/mL. The primary efficacy endpoint was SVR12 (HCV RNA level <25 IU/mL 12 weeks after the last dose of study drug). Secondary efficacy endpoints included (1) the percentage of patients experiencing post-treatment relapse (HCV RNA ≥25 IU/mL within 12 weeks post-treatment in patients who completed treatment with HCV RNA <25 IU/mL) and (2) the percentage of patients experiencing on-treatment virologic failure (failure to achieve HCV RNA <25 IU/mL after 6 weeks of treatment, 2 consecutive HCV RNA measurements that were >1 log10 IU/mL above the nadir at any time point, or 2 consecutive HCV RNA measurements ≥25 IU/mL at any time point after having achieved HCV RNA levels <25 IU/mL). The rates of SVR 4 weeks after the end of treatment (SVR4) and rapid virologic response (RVR; HCV RNA <25 IU/mL at treatment week 4) were also determined.
Resistance Analyses
Resistance testing was performed on all available patient samples at baseline and, for patients who did not achieve SVR, on the first available sample after virologic failure with an HCV RNA level ≥1000 IU/mL. Resistance-associated variants (RAVs) in NS3/4A and NS5A were identified by population sequencing.
Safety
All AEs were recorded from the time of first study drug administration to 30 days after the last dose; serious AEs were monitored throughout the study. All AEs were coded using the Medical Dictionary for Regulatory Activities. The severity of AEs and their relationship to the treatment were assessed by the investigator. Clinical and laboratory parameters were evaluated throughout the study.
Statistical Analyses
A sample size of 40 patients per group was estimated to provide approximately 80% power using a Fisher exact test with a 2-sided significance level of 0.05 to detect a 25% difference between HCV GT1b-infected treatment-naive and prior null responder patients without cirrhosis, assuming that 70% of prior null responders and 95% of treatment-naive patients would achieve SVR12. Efficacy and safety analyses were performed using data from the intent-to-treat population, defined as all patients who received ≥1 dose of study drug. Missing HCV RNA values were imputed using flanking imputation.
The number and percentage of patients achieving efficacy endpoints were summarized, and corresponding exact 95% confidence intervals (CIs) were calculated. SVR rates in patient groups without cirrhosis were compared using a logistic regression model with treatment group, baseline log10 HCV RNA level, and interleukin 28B (IL28B) genotype as predictors; the difference in SVR12 response was assessed using the stratum-adjusted Mantel-Haenszel method adjusted for IL28B genotype. AEs and laboratory values were summarized for each treatment group. SAS/STAT® software (SAS Institute Inc., Cary, NC) for the UNIX operating system was used for all analyses. Statistical tests and 95% CIs were 2-sided with a significance level of 0.05.
Results
Patients
A total of 467 patients with HCV were screened for inclusion in PEARL-I for enrollment in both the GT1b and GT4 arms of the study. Of these patients, 293 had HCV GT1b infection, and 181 (82 noncirrhotic and 99 with compensated cirrhosis) received ≥1 dose of study medication (Supplementary Figure 1). Baseline demographic and clinical characteristics are presented in Table 1. The 2 patient groups without cirrhosis consisted of 42 treatment-naive and 40 prior null responder patients. Of the patients with cirrhosis, 47 were treatment-naive and 52 were pegylated interferon and ribavirin treatment-experienced and had mean baseline platelet count, albumin, and international normalized ratio (PT-INR) of 141.6 x 109 cells/L, 40.4 g/dL, and 1.08, respectively. The vast majority (68.3%-95.0%) of patients had a non-CC IL28B genotype. Nearly half of the treatment-experienced patients with cirrhosis (n = 25 [48.1%]) were prior null responders. Baseline characteristics in treatment-naive and treatment-experienced patients with cirrhosis were similar. Among patients without cirrhosis, a greater percentage of null responder versus treatment-naive patients were female, white, had a non-CC IL28B genotype, or had fibrosis stage F0-F1. The majority of all patients (96.1%) completed treatment; only 7 patients discontinued prematurely (Supplementary Figure 1).
Virologic Response
All but 3 patients achieved RVR at week 4, and SVR12 rates were similarly high in patients with and without cirrhosis (Figure 1). SVR12 was achieved in 95.2% (n = 40/42; 95% CI, 83.8%-99.4%) of treatment-naive and 90.0% (n = 36/40; 95% CI, 76.3%-97.2%) of prior null responder patients without cirrhosis. Among patients with cirrhosis, SVR12 was achieved in 97.9% (n = 46/47; 95% CI, 88.7%-99.9%) of treatment-naive and 96.2% (n = 50/52; 95% CI, 86.8%-99.5%) of treatment-experienced patients. After the primary database lock, 1 treatment-naive patient who was thought to be lost to follow-up returned for a visit and had undetectable HCV RNA levels, making the SVR12 rate following the database lock 100% (n = 47/47) in treatment-naive patients with cirrhosis. No clinically meaningful differences in SVR12 rates were observed between any of these groups. No patient experienced relapse after having achieved SVR12.
Virologic Failure
Five patients experienced virologic failure: 4 prior null responder patients without cirrhosis and 1 treatment-experienced (prior null responder) patient with cirrhosis; all patients had CT IL28B genotype. One patient without cirrhosis experienced on-treatment breakthrough during week 8, and 3 relapsed in post-treatment weeks 3, 5, and 9 (n = 1 each). The patient with cirrhosis relapsed during post-treatment week 2.
Resistance-Associated Variants
Overall, RAVs in NS3 and NS5A were detected in 1.1% and 17.6% of patients at baseline, respectively (Table 2), although many of the NS5A RAVs do not confer resistance to ombitasvir in vitro as single variants. The most commonly detected baseline variant was Y93H in NS5A (n = 13 [7.4%]). In patients without cirrhosis, the SVR12 rate in those with baseline RAVs versus those without RAVs was 92.9% (n = 13/14) versus 96.3% (n = 26/27) in treatment-naive patients and 92.6% (n = 25/27) versus 84.6% (n = 11/13) in prior null responders. In patients with cirrhosis, the SVR12 rate in those with baseline RAVs versus those without RAVs was 96.6% (n = 28/29) versus 100% (n = 18/18) in treatment-naive patients and 96.3% (n = 26/27) versus 95.8% (n = 23/24) in treatment-experienced patients.
No association was observed between specific baseline NS3 and NS5A RAVs and SVR12; the Y93H NS5A variant at baseline was detected in 2 of the 4 prior null responder patients without cirrhosis who did not achieve SVR12 (Y93H, n = 1; P58S + Y93H, n = 1) and it was detected in 2 prior null responder and 2 treatment-naive patients who achieved SVR12. None of the patients with cirrhosis who had RAVs in NS5A at baseline experienced virologic failure. RAVs in NS3 and NS5A were present after breakthrough or relapse in all 5 of the patients with virologic failure (NS3: D168V [n = 4], Y56H + D168A [n = 1]; NS5A: Y93H [n = 3], P58S + Y93H [n = 2]; Table 3).
Safety
Similar rates of treatment-emergent AEs were reported across patient groups (73.1%-80.9%; Table 4), and most were mild in severity. The most commonly reported events were headache (17.3%-33.3%), asthenia (5.0%-21.3%), pruritus (0%-17.0%), and diarrhea (0%-14.9%). Serious AEs occurred in 7 patients (n = 1 each): extrusion of penile prosthesis, chronic obstructive pulmonary disease exacerbation, esophageal variceal hemorrhage, humerus fracture and partial seizures, elevated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels (see the following summary of laboratory abnormalities for further details), peripheral artery aneurysm, and hepatic neoplasm. Only the elevated ALT and AST levels were considered by the investigator to have a reasonable possibility of being related to the study drug. AEs resulted in 3 patients prematurely discontinuing treatment; all 3 patients were treatment-naive with cirrhosis. One of the patients, a 62-year-old male with a previous history of esophageal varices, developed esophageal variceal hemorrhage and prematurely discontinued the study drug on day 24. On day 28 (post-treatment day 4), increased transaminases, renal impairment, and ascites were noted. This event (esophageal variceal hemorrhage) resolved on post-treatment day 17 but the patient subsequently experienced another episode of gastrointestinal hemorrhage (post-treatment day 83) and died 11 days later (post-treatment day 94). The other 2 patients prematurely discontinued study drug because of nonserious events of ascites in the setting of hepatic neoplasm (n = 1) and isolated peripheral edema in the setting of calcium channel blocker use (n = 1).
Laboratory Abnormalities
A grade 2 (<10.0-8.0 g/dL) reduction in hemoglobin occurred in a patient with cirrhosis who had a grade 1 abnormality at baseline. This reduction in hemoglobin persisted throughout the study and required no action with the study drug (Table 4). No grade 3 or 4 reductions in hemoglobin were observed during treatment.
The number of patients with grade 2 or greater elevations in ALT is presented in Table 4. Four patients had grade ≥3 elevations in ALT levels during the treatment period. Two patients (1 with and 1 without cirrhosis) were asymptomatic with no related AEs. A third patient, a 39-year-old female without cirrhosis, required a study drug interruption (days 20-36) because of AEs of ALT, AST, and blood bilirubin increased; the patient resumed study drug on day 37 and achieved SVR12. The fourth patient had cirrhosis and concomitant direct hyperbilirubinemia, edema, and serious AEs of ALT and AST increase on day 45. None of the patients with grade ≥3 elevations in ALT discontinued the study drug, and no interventions were required for these laboratory abnormalities other than study drug interruption in the third patient mentioned above (without cirrhosis); all 4 of these patients achieved SVR12. ALT elevations resolved with continued study treatment by the final visit. Mean ALT and AST changes from baseline are shown in the Supplemental Table. Mean decreases in ALT and AST were observed in all treatment groups and the decreases were generally greater for the patients with cirrhosis than without cirrhosis.
The number of patients with grade 3 elevations in total bilirubin is shown in Table 4. No grade 4 elevations in total bilirubin were observed during treatment. Two patients with cirrhosis (with no ALT/AST elevations) had grade 3 total bilirubin increases that were predominantly indirect and occurred at only 1 visit (days 15 and 112, respectively).
Discussion
Patients with HCV GT1 infection and cirrhosis or those for whom prior treatment with pegylated interferon and ribavirin has failed have historically been difficult to treat successfully. Recent clinical trials have demonstrated that a combination of potent DAAs targeting different stages of the viral life cycle is an efficacious treatment approach for patients with HCV GT1 infection without the use of interferon. In this phase 2b, international, multicenter clinical trial in 181 patients from North America and Europe with HCV GT1b infection, an interferon- and ribavirin-free regimen of ombitasvir, paritaprevir, and ritonavir resulted in high SVR12 rates of 90% to 98% in patients without cirrhosis or with compensated cirrhosis, including patients who were prior null responders. These high SVR12 rates are comparable with or are higher than those reported for other 2-DAA14, 15, 22, 29, 30, 31 and 3-DAA13, 16, 17, 20, 25, 26, 32, 33 interferon-free regimens.
The results described here provide information that was previously lacking in prior null responder patients with GT1b infection. Few studies have evaluated this specific subpopulation and, if they have, efficacy data for such patients are not discernible from the published literature.14, 22, 30, 34, 35 For example, 1 trial that examined the efficacy and safety of sofosbuvir and ledipasvir combined SVR results from patients with heterogeneous treatment histories; efficacy results in specific patient subsets, such as null responders, were not provided.14 One phase 3 study with well-characterized SVR rates by treatment history noted SVR rates of 82% with daclatasvir plus asunaprevir among prior null responders; however, patients with cirrhosis were not evaluated.30 In another trial, only 41 patients with GT1 infection were prior interferon and ribavirin null responders and none had cirrhosis.35
One trial that fully characterized SVR rates by patient subsets examined the safety and efficacy of grazoprevir, an NS3/4A protease inhibitor, and elbasvir, an NS5A protease inhibitor, combined with ribavirin. In this trial, the combination regimen provided SVR12 rates of 90% and 97% with 12 and 18 weeks of treatment, respectively, in treatment-naive patients with HCV GT1 infection without cirrhosis. Similar rates were also observed with the ribavirin-free combination (97% and 94%).22 In the same study, grazoprevir plus elbasvir with or without ribavirin for 12 and 18 weeks also provided high SVR12 rates (91%-100%) in pegylated interferon and ribavirin null responder patients with or without cirrhosis.22 However, the CIs were wide because the number of GT1b-infected cirrhotic patients with a prior null response in this trial was limited (n = 15).22 Another trial that characterized SVR rates by patient subsets was in pegylated-interferon and ribavirin treatment-experienced Japanese patients with HCV GT1b infection without cirrhosis.34 The study evaluated the interferon- and ribavirin-free regimen of ombitasvir (25 mg), paritaprevir (100 or 150 mg), and ritonavir (100 or 150 mg) for 12 or 24 weeks.34 The SVR rates were high (88.9%-100%) and in prior null responder GT1b-infected patients (n = 51), SVR rates were 100% regardless of paritaprevir dose or treatment duration.34 The small differences in SVR between the present and other studies in prior null responder GT1b-infected patients suggest that further studies are needed to determine if this subset of treatment-experienced patients should receive longer treatment duration or an additional DAA.
A low rate (2%; 5/181) of virologic failure was observed in this study; virologic relapse occurred in 4 patients and virologic breakthrough occurred in 1 patient. All 5 patients had RAVs in both NS3 and NS5A at the time of failure, including 2 patients who had RAVs at baseline (both NS5A). It is worth noting that SVR12 rates were similar between patients with and without virus harboring RAVs at baseline (92.9% vs 96.3%); these findings suggest a high resistance barrier for ombitasvir, paritaprevir, and ritonavir in the treatment of HCV GT1b infection. The impact of RAVs at the time of treatment failure on future treatment options is a key topic in the era of interferon-free DAA regimens. A recent analysis in patients with GT1a infection who experienced virologic failure with the 3-DAA regimen of ombitasvir, paritaprevir, and ritonavir plus dasabuvir reported that some NS3 and NS5A RAVs persisted through 48 weeks post-treatment.36 Another report among patients with GT1b infection who failed daclatasvir plus asunaprevir showed that NS5A RAVs remained at high frequency through post-treatment weeks 103 through 170, while NS3 RAVs were replaced by wild-type variants in all patients.37 Together, these data suggest that the persistence of NS5A RAVs is a key factor in evaluating treatment options for patients with GT1 infection who are failing current interferon-free DAA regimens. Studies are needed to evaluate the optimal re-treatment strategy for these patients.
In the PEARL-I trial, treatment with ombitasvir, paritaprevir, and ritonavir was generally well tolerated. Anemia was reported in 1 patient and was not considered related to treatment. The majority of AEs were mild in severity and occurred at similar rates in all groups. There was 1 discontinuation in a cirrhotic patient due to a treatment-related AE (isolated peripheral edema that resolved after study drug discontinuation). Grade 1 or 2 decreases in hemoglobin levels occurred more frequently in patients with cirrhosis than in patients without cirrhosis, but the elevations were mild and did not require any changes in study drug regimen. None of the 4 patients who had grade ≥3 elevations in ALT levels during the treatment period discontinued the study drug, and no interventions were required for the laboratory abnormalities other than study drug interruption in 1 patient. Three patients had grade 3 elevations in total bilirubin; in 2 cases, these elevations were mostly indirect, were not associated with elevated aminotransferases, and were present at only a single visit. These rates are lower than those reported among similar patient populations who received ribavirin, which is known to elevate bilirubin because of hemolysis. Paritaprevir also increases bilirubin levels indirectly through its inhibition of the bilirubin transporter OATP1B1 and may augment the already increased bilirubin levels due to hemolysis.38
It is important to specifically evaluate the efficacy and safety profiles of interferon-free regimens in patients with cirrhosis who have generally had lower SVR rates with interferon-based therapies and who experienced higher rates of AEs than noncirrhotic patients.39, 40, 41, 42 In patients for whom prior pegylated interferon and ribavirin treatment failed, a combination of sofosbuvir and ledipasvir with or without ribavirin for 12 weeks had lower response rates in cirrhotic patients (82%-86%) than noncirrhotic patients (95%-100%).14 When the duration of treatment was increased from 12 to 24 weeks, a significantly greater proportion of patients with cirrhosis (99% with or without ribavirin) achieved SVR12.14 In the present study, an intent-to-treat analysis showed that 97.0% of patients with cirrhosis achieved SVR12 following 24 weeks of treatment with ombitasvir, paritaprevir, and ritonavir. In addition, SVR12 rates in patients with cirrhosis were similar among treatment-naive (97.9%) and treatment-experienced patients (96.2%). Whether a shorter 12-week course of therapy with ombitasvir and paritaprevir may also lead to comparable SVR rates in patients with cirrhosis has not yet been explored. Rates of AEs in patients with cirrhosis were similar to those in patients without cirrhosis, and only 1 patient with cirrhosis discontinued treatment because of an AE related to the study drug. These results suggest that a 24-week course of therapy does not increase the risk of AEs compared with a shorter 12-week course. It is also worth noting that the overall discontinuation rate for any reason was approximately 4% (n = 7/181).
A limitation of this study was that the treatment-experienced group of patients without cirrhosis only included patients who had a null response to prior pegylated interferon and ribavirin treatment; therefore, a full assessment in patients in whom interferon-based treatment has failed is unknown. However, among such patients, null responders are considered the most difficult to treat; the high rates of SVR12 achieved in these patients suggest that ombitasvir, paritaprevir, and ritonavir would be equally efficacious in patients with a prior partial response or relapse. Another limitation is that a 12-week treatment duration was not studied in patients with cirrhosis. Although patients with decompensated cirrhosis were not included in this study, patients with Child-Pugh class B cirrhosis will be evaluated in future clinical trials of ombitasvir, paritaprevir, and ritonavir plus dasabuvir with and without ribavirin.
In conclusion, an all-oral interferon- and ribavirin-free regimen of ombitasvir, paritaprevir, and ritonavir was generally well tolerated and achieved high rates of SVR12 in both cirrhotic and noncirrhotic patients with HCV GT1b infection who were treatment-naive or treatment-experienced, including prior null responders.
|
|
| |
| |
|
|
|