| |
Safety and efficacy of the HIV-1 attachment inhibitor prodrug BMS-663068 in treatment-experienced individuals: 24 week results of AI438011, a phase 2b, randomised controlled trial
|
| |
| |
Download the PDF here
Download the PDF here
The Lancet HIV Oct 2015
Link to very interesting video explaining how the attachment inhibitor blocks HIV:
http://www.thelancet.com/cms/attachment/2037280042/2051782367/mmc3.mp4
"The antiviral activity of BMS-663068 in combination with raltegravir plus TDF was confirmed at the week 24 primary endpoint. The BMS-663068 groups showed similar antiviral efficacy to that of the ritonavir-boosted atazanavir reference group, with a similar proportion of BMS-663068-treated and ritonavir-boosted atazanavir-treated participants in the modified intention-to-treat and observed populations achieving plasma HIV-1 RNA of less than 50 copies per mL at week 24. In a 7 day monotherapy substudy BMS-663068 was active at all doses; for the 800 mg twice daily, 600 mg once daily, and 1200 mg once daily groups, the treatment effect was similar to that of other potent antiretroviral drugs......BMS-663068 is being investigated in a phase 3 study in heavily treatment-experienced, HIV-1-infected individuals.
In the primary efficacy analysis, response rates (proportion of patients with HIV-1 RNA less than 50 copies per mL) at week 24 were similar across all four BMS-663068 groups and the ritonavir-boosted atazanavir comparator group, by analysis with both the FDA-defined snapshot algorithm (modified intention-to-treat population) and observed population criteria (table 2).
According to the modified intention-to-treat snapshot analysis, 69-80% of patients receiving BMS-663068 and 75% of those receiving ritonavir-boosted atazanavir had HIV-1 RNA less than 50 copies per mL. In the observed analysis, 78-87% of patients receiving BMS-663068 and 86% of those receiving ritonavir-boosted atazanavir had HIV-1 RNA less than 50 copies per mL.
Findings from a proof-of-concept study showed a maximum median decrease in plasma HIV-1 RNA viral load from baseline of 1·21-1·73 log10 copies per mL after 8 days of BMS-663068 monotherapy. 42 of 48 patients achieved a decrease in HIV-1 RNA of more than 1·0 log10 copies per mL; however, the decrease of less than 1·0 log10 copies per mL in the remaining six participants (maximum change in HIV-1 RNA from baseline of 0·04 to -0·99 log10 copies per mL) was associated with a baseline BMS-626529 half-maximum inhibitory concentration (IC50) greater than 100 nmol/L.....On the basis of the findings from the proof-of-concept study, we designed this phase 2b dose-finding study to provide an initial assessment of the safety, efficacy, and dose-response of BMS-663068 as part of cART, in HIV-1-infected, treatment-experienced patients who had received at least 1 week of previous antiretroviral therapy. We present the findings at 24 weeks.
BMS-626529 is given as the prodrug BMS-663068 to overcome its solubility-limited bioavailability.....BMS-626529 is an HIV-1 attachment inhibitor that prevents the initial interaction between the virus and the host cell by binding to the viral envelope protein gp120 (see online video S1). This binding blocks the attachment of the virus to the CD4 receptor of CD4 cells. By targeting the initial step of viral attachment, before co-receptor binding and fusion, BMS-626529 is not affected by co-receptor tropism, and is active against C-C chemokine receptor type 5 (CCR5), C-X-C chemokine receptor type 4 (CXCR4), and dual-tropic (R5X4) strains of HIV-1. Preliminary in-vitro data show that HIV-1 viruses are generally susceptible to BMS-626529, with the exception of subtype AE and group O; however, no activity was shown against a strain of HIV-2. BMS-626529 also has a unique resistance profile, and cross-resistance to other classes of antiretroviral drugs is not observed in vitro.
On the basis of these results, and because there is a significant need for new antiretroviral drugs for treatment-experienced patients who have limited remaining treatment options due to resistance, toxic effects, or drug interactions, BMS-663068 is being investigated in a phase 3 study in heavily treatment-experienced, HIV-1-infected individuals. The study will not include any restrictions on baseline BMS-626529 IC50 and a retrospective exploratory analysis will be done to determine the effect of baseline BMS-626529 IC50 on antiviral response. The results of the phase 3 study, combined with ongoing work to determine the genotypic and phenotypic correlates of susceptibility to BMS-626529, will be used to determine whether or not a diagnostic assay will be required for use of BMS-663068.
Eligible participants were at least 18 years of age, treatment-experienced (current or previous exposure to 1 week or more of at least one antiretroviral drug) with a plasma HIV-1 RNA viral load of at least 1000 copies per mL and a CD4 cell count greater than 50 cells per μL. Participants had to have less than 1 week of previous exposure to any integrase inhibitor and a screening drug-resistance genotype and phenotype indicating susceptibility to ritonavir-boosted atazanavir, raltegravir, and TDF. To ensure maximum efficacy in this early treatment-experienced population, and on the basis of the results from the proof-of-concept study,13, 16 participants also had to have a BMS-626529 IC50 of less than 0·1 μmol/L (100 nmol/L). Detailed eligibility criteria are provided in the study protocol (appendix 2)."
--------------------
Safety and efficacy of the HIV-1 attachment inhibitor prodrug BMS-663068 in treatment-experienced individuals: 24 week results of AI438011, a phase 2b, randomised controlled trial
The Lancet HIV
Sept 2 2015
Jacob P Lalezari, Gulam H Latiff , Cynthia Brinson, Juan Echevarr�a, Sandra Trevino-Perez, Johannes R Bogner, Melanie Thompson, Jan Fourie,
Otto A Sussmann Pena, Fernando C Mendo Urbina, Marcelo Martins, Iulian G Diaconescu, David A Stock, Samit R Joshi, George J Hanna,
Max Lataillade, for the AI438011 study team
Summary
Background
BMS-663068 is an oral prodrug of BMS-626529, an attachment inhibitor that binds to HIV-1 gp120, blocking viral attachment to host CD4 cells. AI438011 is an ongoing trial investigating the efficacy, safety, and dose-response of BMS-663068 in treatment-experienced, HIV-1-infected patients. Herein we present the results of the primary analysis.
Methods
AI438011 is a phase 2b, randomised, active-controlled trial, at 53 hospitals and outpatient clinics across ten countries in North and South America, Europe, and Africa. Individuals with an HIV-1 RNA viral load of at least 1000 copies per mL and a BMS-626529 half-maximum inhibitory concentration lower than 100 nmol/L were randomly assigned (1:1:1:1:1) to receive either BMS-663068 at 400 mg twice daily, 800 mg twice daily, 600 mg once daily, or 1200 mg once daily or ritonavir-boosted atazanavir (300 mg of atazanavir and 100 mg of ritonavir once daily), each with 400 mg of raltegravir twice daily and 300 mg of tenofovir disoproxil fumarate once daily as a backbone. The sponsor, participants, and investigators were masked for BMS-663068 dose but not for allocation. Primary endpoints were the proportion of patients with an HIV-1 RNA viral load less than 50 copies per mL (response rate) at week 24 and the frequency of serious adverse events and adverse events leading to discontinuation, up to the week 24 analysis. The primary analyses included all patients who received at least one dose of study drug (modified intention-to-treat population). This study is registered at ClinicalTrials.gov, NCT01384734.
Findings
Between July 26, 2011, and July 16, 2012, 581 participants were assessed for eligibility. Of these, 254 patients were randomly assigned to receive either BMS-663068 (n=52 for the 400 mg twice daily group, n=50 for the 800 mg twice daily group, n=51 for the 600 mg once daily group, and n=50 for the 1200 mg once daily group) or ritonavir-boosted atazanavir (n=51). 200 patients received at least one dose of BMS-663068, and 51 patients received at least one dose of ritonavir-boosted atazanavir. At week 24, 40 (80%) of 50 patients in the BMS-663068 400 mg twice daily group, 34 (69%) of 49 patients in the 800 mg twice daily group, 39 (76%) of 51 patients in the 600 mg once daily group, and 36 (72%) of 50 patients in the 1200 mg once daily group had an HIV-1 RNA viral load less than 50 copies per mL, compared with 38 (75%) of 51 patients in the ritonavir-boosted atazanavir group. Serious adverse events were noted in 13 (7%) of 200 patients in the BMS-663068 groups and five (10%) of the 51 patients in the ritonavir-boosted atazanavir group. Four (2%) of the 200 patients in the BMS-663068 groups and two (4%) of the 51 patients in the ritonavir-boosted atazanavir group discontinued because of adverse events. No serious adverse events or adverse events leading to discontinuation were BMS-663068-related. Grade 2-4 adverse events related to study drug(s) occurred in 17 (9%) of 200 patients across the BMS-663068 groups and 14 (27%) of 51 patients in the ritonavir-boosted atazanavir group. For the BMS-663068 groups these events were mostly single instances with no dose relation and for the ritonavir-boosted atazanavir group these were mostly gastrointestinal or hepatobiliary disorders associated with hyperbilirubinaemia.
Interpretation
In a comparison with ritonavir-boosted atazanavir, efficacy and safety of BMS-663068 up to the week 24 analysis support continued development of BMS-663068, which is being assessed in a phase 3 trial in heavily treatment-experienced individuals.
Funding
Bristol-Myers Squibb.
Introduction
Treatment of HIV-1-infection requires lifelong treatment with combination antiretroviral therapy (cART). Despite substantial improvements in life expectancy,1, 2 virological failure still occurs3, 4, 5 and treatment modifications are common.6, 7 New treatment regimens should preferably include three fully active drugs, selected on the basis of a patient's treatment history and assessment of adherence, drug interactions, tolerability, and current or historical viral drug resistance patterns.8, 9 Thus, there is a constant need for antiretroviral drugs with new mechanisms of action that can be successfully combined with other drugs to construct suppressive, well tolerated regimens for use in later lines of therapy.
Research in context
Evidence before this study
We searched PubMed on March 18, 2015, using the search terms "HIV Attachment Inhibitor", "BMS-663068", and "BMS-626529" with no date restrictions, and we found two previous clinical studies of HIV-1 attachment inhibitors. One study showed the efficacy of a previous HIV-1 attachment inhibitor candidate during 8 days of monotherapy; and the other reported the findings of a phase 2a study, investigating the pharmacokinetics, pharmacodynamics, and safety of BMS-663068 after 8 days of monotherapy. In the BMS-663068 phase 2a study, 8 days of monotherapy in HIV-1-infected patients resulted in substantial decreases in plasma HIV-1 RNA viral load (1·21-1·73 log10 copies per mL) and BMS-663068 was generally well tolerated. Additionally, no emergent changes in viral susceptibility were noted over the course of the study according to population phenotypic and genotypic approaches. These data were used to design this phase 2b study.
Added value of this study
AI438011 is the first study to investigate safety and efficacy of an HIV-1 attachment inhibitor as part of combination antiretroviral therapy. Data from the phase 2b, week 24 primary endpoint analysis show that BMS-663068 has similar efficacy to ritonavir-boosted atazanavir when combined with a backbone of raltegravir and tenofovir disoproxil fumarate in a treatment-experienced population. BMS-663068 was generally well tolerated throughout the study, and no BMS-663068-related serious adverse events or adverse events leading to discontinuation were observed.
Implications of all the available evidence
Attachment inhibitors are a new class of antiretroviral drugs. By binding directly to the viral envelope protein, gp120, they inhibit binding of the virus to CD4 and act before the co-receptor binding and membrane fusion steps targeted by CCR5 antagonists and fusion inhibitors, respectively. As such, attachment inhibitors can be used to target CCR5-tropic, CXCR4-tropic, and dual-tropic strains of HIV-1. Additionally, the unique resistance profile and absence of in vitro cross-resistance observed with other classes of antiretroviral drugs suggests that they might be of particular benefit for individuals with extensive viral drug resistance. On the basis of our findings, BMS-663068 is being assessed in a phase 3 study in heavily treatment-experienced individuals, for whom there is an unmet need for new antiretroviral drugs.
BMS-626529 is an HIV-1 attachment inhibitor that prevents the initial interaction between the virus and the host cell by binding to the viral envelope protein gp120 (see online video S1). This binding blocks the attachment of the virus to the CD4 receptor of CD4 cells.10 By targeting the initial step of viral attachment, before co-receptor binding and fusion, BMS-626529 is not affected by co-receptor tropism, and is active against C-C chemokine receptor type 5 (CCR5), C-X-C chemokine receptor type 4 (CXCR4), and dual-tropic (R5X4) strains of HIV-1.10, 11, 12, 13, 14 Preliminary in-vitro data12 show that HIV-1 viruses are generally susceptible to BMS-626529, with the exception of subtype AE and group O; however, no activity was shown against a strain of HIV-2.12 BMS-626529 also has a unique resistance profile, and cross-resistance to other classes of antiretroviral drugs is not observed in vitro.11, 12
BMS-626529 is given as the prodrug BMS-663068 to overcome its solubility-limited bioavailability. Alkaline phosphatase converts the prodrug to the active agent in the gastrointestinal tract immediately before absorption (appendix 1, p 2).15 Findings from a proof-of-concept study16 showed a maximum median decrease in plasma HIV-1 RNA viral load from baseline of 1·21-1·73 log10 copies per mL after 8 days of BMS-663068 monotherapy. 42 of 48 patients achieved a decrease in HIV-1 RNA of more than 1·0 log10 copies per mL; however, the decrease of less than 1·0 log10 copies per mL in the remaining six participants (maximum change in HIV-1 RNA from baseline of 0·04 to -0·99 log10 copies per mL)17 was associated with a baseline BMS-626529 half-maximum inhibitory concentration (IC50) greater than 100 nmol/L.13, 16
On the basis of the findings from the proof-of-concept study,13, 16 we designed this phase 2b dose-finding study to provide an initial assessment of the safety, efficacy, and dose-response of BMS-663068 as part of cART, in HIV-1-infected, treatment-experienced patients who had received at least 1 week of previous antiretroviral therapy. We present the findings at 24 weeks.
The results of this phase 2b study contributed to the selection of a dose for a registrational phase 3 study (ClinicalTrials.gov, NCT02362503). This phase 3 trial is being done in a heavily treatment-experienced population who have limited therapeutic options.
Methods
Study design and participants
AI438011 is a phase 2b, randomised, active-controlled, blinded-to-BMS-663068-dose trial (appendix 1, p 2) done at 53 hospitals and outpatient clinics across ten countries in South America, North America, Europe, and Africa. The accompanying study protocol is available with the full-text article online (appendix 2).
HIV-1 treatment guidelines do not specify a standard-of-care regimen for infected individuals for whom first-line and subsequent treatment regimens have failed.8, 9 We selected a fixed backbone of raltegravir and tenofovir disoproxil fumarate (TDF) because it was anticipated that most screened participants would have a virus susceptible to both drugs, and, therefore, combined with a third agent this backbone would provide participants with a fully active treatment regimen. Additionally, this fixed backbone allowed for BMS-663068 dose differentiation for subsequent clinical development. A reference arm of ritonavir-boosted atazanavir combined with the backbone raltegravir and TDF provided an active comparator. A subset of participants (up to ten participants per BMS-663068 group) also took part in a 7 day elective monotherapy substudy before their participation in the main study to confirm the antiretroviral activity of BMS-663068. An independent data monitoring committee did an interim analysis at week 8 to assess early virological efficacy and safety endpoints.
The study was done in accordance with international laws and guidelines and designed and conducted according to the protocol (and any amendments) jointly by the study investigators and sponsor. Each site conducted the protocol with overview and approval from an Institutional Review Board or Independent Ethics Committee.
Eligible participants were at least 18 years of age, treatment-experienced (current or previous exposure to 1 week or more of at least one antiretroviral drug) with a plasma HIV-1 RNA viral load of at least 1000 copies per mL and a CD4 cell count greater than 50 cells per μL. Participants had to have less than 1 week of previous exposure to any integrase inhibitor and a screening drug-resistance genotype and phenotype indicating susceptibility to ritonavir-boosted atazanavir, raltegravir, and TDF. To ensure maximum efficacy in this early treatment-experienced population, and on the basis of the results from the proof-of-concept study,13, 16 participants also had to have a BMS-626529 IC50 of less than 0·1 μmol/L (100 nmol/L). Detailed eligibility criteria are provided in the study protocol (appendix 2).
All participants provided written informed consent in agreement with the Declaration of Helsinki principles.
Randomisation and masking
Eligible patients were randomly assigned (1:1:1:1:1) to five treatment groups: four BMS-663068 groups (with doses of 400 mg twice daily, 800 mg twice daily, 600 mg once daily, and 1200 mg once daily), and a control group given ritonavir-boosted atazanavir (300 mg of atazanavir with 100 mg of ritonavir once daily). All treatments were given with a backbone of raltegravir 400 mg twice daily and TDF 300 mg once daily. Randomisation was done with a computer-generated code and stratified by phenotypic sensitivity to BMS-626529 (IC50 <1·3 nmol/L or ≥1·3 nmol/L), measured with the Monogram Biosciences PhenoSense Entry assay. Participants and investigators were masked to BMS-663068 doses, but not to allocation. Unmasking occurred after the last participant reached week 24 for the sponsor or week 48 for the participants and investigators.
Procedures
Approximately ten randomised patients per treatment group opted in to participate in the monotherapy substudy for 7 days. All treatments were taken as oral tablets. Study visits were completed at screening, on days 1, 2, 5, 6, and 7 during the monotherapy substudy, and on day 1 and weeks 2, 4, 8, 12, 16, 20, 24 (or on early termination) in the main study. Patients participating in the monotherapy substudy began dosing for the main study the day after they received the final monotherapy dose. We quantified plasma HIV-1 RNA with the Roche COBAS Amplicor assay (Roche Molecular Diagnostics, California, USA). Protocol-defined virological failure (PDVF) was confirmed HIV-1 RNA of at least 50 copies per mL at week 24, or later (second measurement within 2-4 weeks of original sample), or virological rebound (confirmed HIV-1 RNA ≥50 copies per mL at any time after previous confirmed suppression to <50 copies per mL, or confirmed increase in HIV-1 RNA of ≥1 log10 copies per mL above the nadir level at any time when the nadir is ≥50 copies per mL). In the event of virological failure with a confirmatory viral load measurement of less than 1000 copies per mL, and in the absence of emergent viral drug resistance, participants could remain on-study at the discretion of the investigator to week 48. Those with a confirmatory HIV-1 RNA measurement greater than or equal to 1000 copies per mL were withdrawn from the study after week 24. CD4 cell counts were measured on day 1 of the monotherapy substudy, and throughout the main study.
Safety assessments, including vital signs and physical examination, adverse events, laboratory measurements, and electrocardiographs were recorded throughout the main study. Adverse events were coded according to MedDRA version 15.1. Investigators assessed the severity of adverse events and their relation to the study drug.
Viral drug resistance was tested at screening and in the event of confirmed PDVF, or at a minimum in the event of a confirmed HIV-1 RNA viral load measurement of at least 400 copies per mL at any time during the study (having previously achieved viral suppression to <50 copies per mL), or discontinuation before achieving viral suppression after week 8 with a last HIV-1 RNA measurement of at least 400 copies per mL. Genotypic and phenotypic nucleos(t)ide reverse transcriptase inhibitor (NRTI), non-NRTI (NNRTI) and protease inhibitor resistance, and integrase inhibitor resistance were determined for screening and confirmatory samples with the Monogram Biosciences PhenoSense GT and Integrase (GeneSeq and PhenoSense) assays (LabCorp, South San Francisco, CA, USA). We analysed viral susceptibility to BMS-626529 with the Monogram Biosciences PhenoSense Entry assay (LabCorp, South San Francisco, CA, USA) and reported it as fold-change in IC50 versus a reference virus (DUAL) with an IC50 of about 1 nmol/L (range 0·5-1·9) that was used for normalisation, as previously reported.13 We used a greater than three-fold cutoff for analysis of changes in BMS-626529 fold-change in IC50 from baseline, because at least 95% of replicate fold-change in IC50 measurements in the PhenoSense Entry assay are within three-fold of each other.13
The study is ongoing. We present the findings at 24 weeks, and the results of long-term follow-up through secondary endpoints at weeks 48 and 96 will be presented elsewhere.
Outcomes
Primary endpoints were proportion of participants with plasma HIV-1 RNA viral load less than 50 copies per mL at week 24, frequency of serious adverse events, and adverse events leading to discontinuation up to the week 24 analysis.
Secondary endpoints included assessment of the antiviral activity of BMS-663068 (change in viral load from baseline to day 8 [log10 copies per mL]) in the monotherapy substudy, change from baseline in CD4 cell count and assessment of emergent drug resistance in virus from participants experiencing virological failure up to the week 24 analysis.
We did a prespecified subgroup analysis to assess the proportion of participants achieving an HIV-1 RNA viral load less than 50 copies per mL according to the predefined BMS-626529 IC50 strata (<1·3 nmol/L or ≥1·3 nmol/L), and a series of additional BMS-626529 IC50 breakpoints (<0·1 or ≥0·1 nmol/L, <1·0 or ≥1·0 nmol/L, and <10 or ≥10 nmol/L) spanning the range of baseline IC50 values included in the study. Additionally, we did post-hoc analyses to assess the proportion of participants with plasma HIV-1 RNA less than 50 copies per mL according to participation in the monotherapy substudy or baseline HIV-1 RNA (<100 000 or ≥100 000 copies per mL).
Statistical analysis
This study was an estimation study not powered to show statistical differences between treatment groups. The primary efficacy endpoint assessment used the US Food and Drug Administration (FDA)-defined snapshot algorithm, taking the last on-treatment HIV-1 RNA viral load in the predefined week 24 visit window (±6 weeks) to determine response. The modified intention-to-treat population consisted of randomised participants who received at least one dose of BMS-663068 or ritonavir-boosted atazanavir. The observed population consisted of participants who received at least one dose of BMS-663068 or ritonavir-boosted atazanavir, with HIV-1 RNA measurements within the week 24 window. Additional prespecified and ad-hoc analyses were done according to baseline HIV-1 RNA and BMS-626529 IC50 category.
We selected the target sample size of 50 participants per treatment group to provide the following exact binomial 95% CIs for observed response rates (proportion of participants with HIV-1 RNA less than 50 copies per mL at week 24): response rate of 90% (78-97), 80% (66-90), and 70% (55-82).
This trial is registered with ClinicalTrials.gov (NCT01384734) and the European Clinical Trials Database (EudraCT, 2011-000437-36).
Role of the funding source
The funder of the study participated in the study design, data collection, data analysis, and data interpretation. All authors had full access to the data and vouch for the completeness and accuracy of the data and analyses presented and for the fidelity of the study to the protocol. The first draft of the manuscript was prepared by a professional medical writer, paid for by the funder, and edited and revised by all authors. The corresponding author had final responsibility for the decision to submit for publication.
Results
Between July 26, 2011, and July 16, 2012, 581 participants were screened. Of these, 254 were randomly assigned to treatment groups in the study, and 251 received treatment (figure 1). The last patient received first treatment on Aug 29, 2012. Most screen failures were because of failure to meet study entry criteria; the most common reason was an HIV-1 RNA less than 1000 copies per mL (95 [29%] of the 327 excluded). Of the 435 patients who had a successful PhenoSense Entry assay screening result, about 26 (6%) had a BMS-626529 IC50 greater than 100 nmol/L at baseline. The last patient reached week 24 on Feb 13, 2013, and the week 24 database lock was complete on April 16, 2013. Of those receiving treatment, 32 (16%) of 200 patients in the BMS-663068 groups and nine (18%) of 51 patients in the ritonavir-boosted atazanavir group did not complete 24 weeks of treatment (figure 1). 32 (16%) of the 203 patients randomly assigned to the BMS-663068 groups chose to participate in the BMS-663068 monotherapy substudy. After an interim review by an independent data monitoring committee at week 8, the main study continued as planned without modification.
Baseline demographic and disease characteristics were broadly similar across all treatment groups (table 1). The median age of the participants was 39 years (IQR 31-46), and 151 (60%) of 251 patients were male. Most patients (165 [66%]) had HIV-1 subtype B. Median HIV-1 RNA viral load at baseline was 4·85 log10 copies per mL (IQR 4·24-5·43) and 107 (43%) patients had 100 000 copies per mL or more; median CD4 cell count was 229·5 cells per μL (IQR 153-324) and 96 (38%) had less than 200 cells per μL. Across the groups, 38-61% of patients had at least one major mutation associated with protease inhibitor, NRTI, or NNRTI resistance at baseline; the most commonly observed mutations were the Met184Val (in 22-40% of patient samples) and Lys103Asn (in 20-39% of patient samples) substitutions (appendix 1, p 3).
In the monotherapy substudy, treatment with BMS-663068 monotherapy for 7 days resulted in median changes in HIV-1 RNA from baseline of -0·69 log10 copies per mL (IQR -1·17 to -0·38) for the 400 mg twice daily group, -1·40 log10 copies per mL (-1·59 to -1·16) for the 800 mg twice daily group, -1·28 log10 copies per mL (-1·49 to -0·93) for the 600 mg once daily group, and -1·44 log10 copies per mL (-1·61 to -1·06) for the 1200 mg once daily group (figure 2).
In the primary efficacy analysis, response rates (proportion of patients with HIV-1 RNA less than 50 copies per mL) at week 24 were similar across all four BMS-663068 groups and the ritonavir-boosted atazanavir comparator group, by analysis with both the FDA-defined snapshot algorithm (modified intention-to-treat population) and observed population criteria (table 2). According to the modified intention-to-treat snapshot analysis, 69-80% of patients receiving BMS-663068 and 75% of those receiving ritonavir-boosted atazanavir had HIV-1 RNA less than 50 copies per mL. In the observed analysis, 78-87% of patients receiving BMS-663068 and 86% of those receiving ritonavir-boosted atazanavir had HIV-1 RNA less than 50 copies per mL.
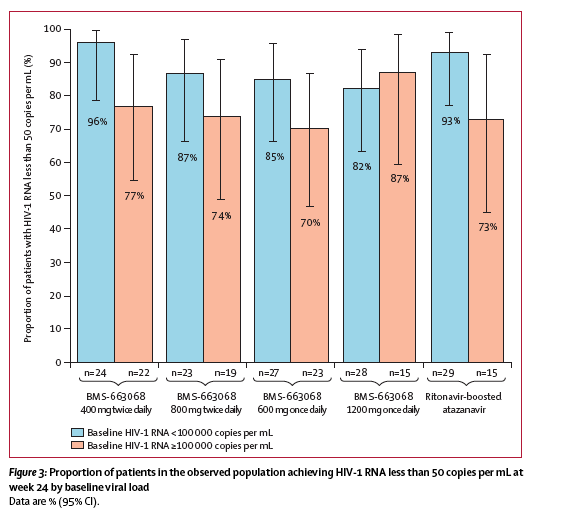
When assessed according to baseline HIV-1 RNA, response rates for patients with a baseline HIV-1 RNA of less than 100 000 copies per mL were better than those for patients with a baseline HIV-1 RNA of at least 100 000 copies per mL (observed analysis); however, response did not seem to differ across the BMS-663068 and ritonavir-boosted atazanavir groups in either group at week 24 (figure 3). Within the predefined baseline BMS-626529 IC50 strata (<1·3 nmol/L and ≥1·3 nmol/L), the proportion of patients with a BMS-626529 IC50 of less than 1·3 nmol/L who achieved response varied from 75% to 92% in the BMS-663068 groups and was 90% in the ritonavir-boosted atazanavir group. The proportion of patients with a BMS-626529 IC50 of 1·3 nmol/L or more who achieved response varied from 65% to 88% in the BMS-663068 groups and was 77% in the ritonavir-boosted atazanavir group (table 3). Additionally, response rates at week 24 were similar across all study groups, across the additional baseline BMS-626529 IC50 breakpoints assessed (table 3).
For patients who participated in the BMS-663068 monotherapy substudy before the main study, seven (100%) of seven patients in the 400 mg twice daily group, four (100%) of four patients in the 800 mg twice daily group, nine (90%) of ten patients in the 600 mg once daily group, and eight (89%) of nine patients in the 1200 mg once daily group had HIV-1 RNA less than 50 copies per mL at week 24 (observed analysis). Response rates observed for patients who did not participate in the monotherapy substudy were 33 (85%) of 39 in the 400 mg twice daily group, 30 (79%) of 38 in the 800 mg twice daily group, 30 (75%) of 40 in the 600 mg once daily group, and 28 (82%) of 34 in the 1200 mg once daily group.
Change from baseline in CD4 cell count at week 24 was similar across the BMS-663068 and ritonavir-boosted atazanavir groups (appendix 1, p 4); however, the median increase in CD4 cell count from baseline seemed to be greater in the 400 mg twice daily group than in any other group. Median increases in CD4 cell count from baseline were 138 cells per μL (IQR 85-192) for the 400 mg twice daily group, 116 cells per μL (52-206) for the 800 mg twice daily group, 100 cells per μL (65-154) for the 600 mg once daily group, 127 cells per μL (71-163) for the 1200 mg once daily group, and 103 cells per μL (35-184) for the ritonavir-boosted atazanavir group.
At the week 24 analysis, 23 (12%) of 200 patients across the BMS-663068 groups and eight (16%) of 51 patients in the ritonavir-boosted atazanavir group met on-study criteria for resistance testing. 21 (91%) of the 23 patients in the BMS-663068 groups and all eight patients in the ritonavir-boosted atazanavir group had samples that were successfully tested using the PhenoSense GT or Integrase assays. No samples showed resistance mutations associated with TDF or atazanavir. Viral susceptibility to BMS-626529 was evaluable with the PhenoSense Entry assay for 19 (83%) of 23 patients. Eight patients had virus that exhibited a greater than three-fold change in BMS-626529 fold-change in IC50 from baseline, and four of eight also had virus exhibiting emergent raltegravir resistance (appendix 1, p 5). No resistance to study drugs was detected in the ritonavir-boosted atazanavir group.
BMS-663068 was generally well tolerated. Across all study groups, six (2%) of 251 patients discontinued because of adverse events (table 4; appendix 1, p 6); however, no BMS-663068-related adverse events led to discontinuation.
The most commonly reported adverse event across the BMS-663068 groups was headache (mostly grade 1), which occurred in 28 (14%) of 200 patients in the BMS-663068 groups compared with five (10%) of 51 patients in the ritonavir-boosted atazanavir group. Grade 2-4 treatment-related adverse events (table 4, appendix 1, p 7) occurred in 17 (9%) 200 patients across the BMS-663068 groups; most were single instances and there was no dose-relationship. In the ritonavir-boosted atazanavir group, 14 (27%) of 51 patients had grade 2-4 treatment-related adverse events, which were mostly gastrointestinal and hepatobiliary disorders associated with hyperbilirubinaemia.
Serious adverse events were reported in 13 (7%) of 200 patients across the BMS-663068 groups and five (10%) of 51 in the ritonavir-boosted atazanavir group. Most of them were attributable to infections and none were related to BMS-663068 or ritonavir-boosted atazanavir (table 4, appendix 1, p 10). No deaths occurred.
Grade 3-4 laboratory abnormalities across the BMS-663068 groups (neutropenia in five [3%] of 200 patients) and increases in aspartate aminotransferase/alanine aminotransferase concentrations (four [2%] of 200) were uncommon with no noticeable trends (data not shown). In the ritonavir-boosted atazanavir group, increased total bilirubin concentration was the most notable Grade 3-4 laboratory abnormality (in 25 [49%] of 51 patients).
Discussion
The antiviral activity of BMS-663068 in combination with raltegravir plus TDF was confirmed at the week 24 primary endpoint. The BMS-663068 groups showed similar antiviral efficacy to that of the ritonavir-boosted atazanavir reference group, with a similar proportion of BMS-663068-treated and ritonavir-boosted atazanavir-treated participants in the modified intention-to-treat and observed populations achieving plasma HIV-1 RNA of less than 50 copies per mL at week 24. In a 7 day monotherapy substudy BMS-663068 was active at all doses; for the 800 mg twice daily, 600 mg once daily, and 1200 mg once daily groups, the treatment effect was similar to that of other potent antiretroviral drugs.18, 19
Immune reconstitution was similar across the BMS-663068 and ritonavir-boosted atazanavir groups at week 24, with median increase in CD4 cell counts in the range of 100-138 cells per μL. Consistent with findings from other trials of antiretroviral combinations,3 higher response rates were achieved by patients with baseline HIV-1 RNA of less than 100 000 copies per mL compared with those who had a baseline HIV-1 RNA greater than or equal to 100 000 copies per mL; however, response rates were similar across the BMS-663068 and ritonavir-boosted atazanavir groups irrespective of baseline HIV-1 RNA category. Response rates did not seem to be affected by participation in the monotherapy substudy.
Ritonavir-boosted atazanavir was selected as a comparator for the study because it is frequently used in cART regimens for treatment-experienced adults and it provided a potent reference group for efficacy comparisons. We selected a standard raltegravir and TDF backbone since both drugs have well defined safety profiles, it allowed for dose differentiation for subsequent clinical development, and most screened patients were likely to have virus susceptible to both drugs, allowing for a fully active treatment backbone for use in cART. The combination of ritonavir-boosted atazanavir, raltegravir, and TDF provided treatment-experienced participants with three fully active drugs constituting a potent cART regimen.
We applied a maximum baseline BMS-626529 IC50 of 100 nmol/L (according to the PhenoSense Entry assay) for entry into the study, based on a reduced maximal antiviral response seen in a small number of individuals in the phase 2a study, in which six of seven participants with a baseline BMS-626529 IC50 of greater than 100 nmol/L had a decrease in HIV-1 RNA viral load of less than 1 log10 copies per mL after 8 days of BMS-663068 monotherapy.13, 16 When assessed according to a range of baseline BMS-626529 IC50 breakpoints, response rates remained similar across the BMS-663068 groups at week 24, suggesting that, within the context of this study, baseline BMS-626529 IC50 did not affect antiviral response. Preliminary data have also shown that a BMS-626529 concentration of 100 nmol/L is expected to exceed the IC50 for 99% of HIV-1 subtype B, 94% of subtype C, and 90% of subtype A viruses based on in-vitro findings.12 On the basis of these results, further investigation will be required to determine the effect of a wider range in baseline viral susceptibility to BMS-626529 on antiviral efficacy, and the relevance of including a diagnostic assay before initiation of therapy.
At the week 24 analysis, 16% of participants across the BMS-663068 groups and 18% in the ritonavir-boosted atazanavir group discontinued from the study; however, most discontinuations were not related to safety or efficacy (all reasons for discontinuation were assessed by the investigators). There was a fairly high pill burden in the study; patients across the BMS-663068 groups took nine pills a day before unblinding and participants in the ritonavir-boosted atazanavir group took five pills a day throughout the study. This high pill burden might have affected adherence; however, this effect was not formally assessed.
BMS-663068 was generally well tolerated at all doses assessed. Headache was the most commonly reported adverse event across the BMS-663068 groups but was mostly grade 1, and not dose-related. There were no safety signals (clinical or laboratory based) across the BMS-663068 groups and no BMS-663068-related serious adverse events or adverse events leading to discontinuation. The most common adverse events and laboratory abnormalities encountered after administration of ritonavir-boosted atazanavir were gastrointestinal and hepatobiliary disorders associated with hyperbilirubinaemia, consistent with the known profile of atazanvir.20
The incidence of virological failure was similar across the BMS-663068 (16-26%) and ritonavir-boosted atazanavir groups (18%), based on the FDA snapshot algorithm. Of the patients meeting resistance testing criteria that were successfully tested, more patients developed treatment-emergent genotypic and phenotypic changes in viral drug susceptibility in the BMS-663068 groups than in the ritonavir-boosted atazanavir group, with eight participants across the BMS-663068 groups having virus with a greater than three-fold increase in BMS-626529 fold-change in IC50 from baseline, and four also having virus with emergent raltegravir resistance-associated mutations. At present, no clinical cutoff for changes in viral susceptibility to BMS-626529 has been defined; the greater than three-fold cutoff used to assess increases in BMS-626529 fold-change in IC50 from baseline was conservative and based on the limits for assay variability. Whether these changes in viral susceptibility to BMS-626529 are clinically relevant is unknown; however, data suggest that BMS-626529 has a lower barrier to resistance than does ritonavir-boosted atazanavir.
Our study had several strengths. The main study population was quite well balanced in terms of baseline variables likely to influence response, including HIV-1 RNA, CD4 cell count, and BMS-626529 IC50. The study was done across four continents, and female participants were well represented. Additionally, about a third of participants had baseline CD4 cell counts of less than 200 cells per μL, and the baseline genotypic resistance profile was illustrative of a treatment-experienced population, with at least 50% of participants having one or more major NRTI, NNRTI, or protease inhibitor resistance-associated mutation at baseline.
The study had some limitations. First, the study population was largely reflective of first-line or second-line virological failure, so further studies will be required to show the antiviral efficacy of BMS-663068 in treatment-experienced populations receiving later lines of therapy, for whom new agents with activity against drug-resistant HIV-1 are needed. Second, we only included participants with a BMS-626529 IC50 less than 100 nmol/L at entry; the efficacy of BMS-663068 (combined with other antiretroviral drugs) in populations that could potentially have higher baseline BMS-626529 IC50 values requires further investigation.
To our knowledge, this is the first study to explore the efficacy of an attachment inhibitor (a novel antiretroviral class) relative to a boosted protease inhibitor, as part of cART. On the basis of these results, and because there is a significant need for new antiretroviral drugs for treatment-experienced patients who have limited remaining treatment options due to resistance, toxic effects, or drug interactions, BMS-663068 is being investigated in a phase 3 study in heavily treatment-experienced, HIV-1-infected individuals. The study will not include any restrictions on baseline BMS-626529 IC50 and a retrospective exploratory analysis will be done to determine the effect of baseline BMS-626529 IC50 on antiviral response. The results of the phase 3 study, combined with ongoing work to determine the genotypic and phenotypic correlates of susceptibility to BMS-626529, will be used to determine whether or not a diagnostic assay will be required for use of BMS-663068.
|
|
| |
| |
|
|
|