 |
|
|
| |
Ledipasvir and Sofosbuvir for HCV in Patients Coinfected with HIV-1
|
| |
| |
Download the PDF here
Download the PDF here
July 21 2015 NEJM
"None of the 335 patients in the study discontinued treatment prematurely because of an adverse event"
"In this multicenter, open-label, single-group study, 12 weeks of treatment with the once-daily, single-tablet regimen of ledipasvir-sofosbuvir resulted in a sustained virologic response in 96% of patients. ....
......In exploratory subgroup analyses, rates of sustained virologic response 12 weeks after the end of therapy (the primary efficacy end point) were similar across all subgroups except that black patients, who made up 34% of the study population, had lower rates of sustained virologic response. This association between black race and a decreased rate of virologic response was not observed in the 308 black patients who were monoinfected with HCV receiving ledipasvir-sofosbuvir across the phase 3 program.23-25 The CYP2B6 polymorphism, which is more common among blacks and has been reported to be associated with higher serum efavirenz levels, was assessed in a candidate-gene analysis and was not associated with relapse.26 A genomewide association study might be able to identify genetic factors associated with this observation."
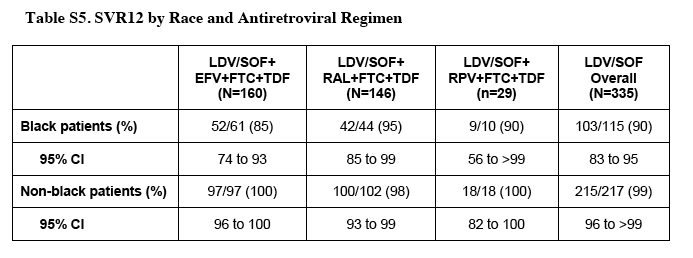
"In total, 13 patients (4%) did not have a sustained virologic response. Of these patients, 1 died after 4 weeks of treatment, 2 had HCV breakthrough during treatment that was associated with suspected poor adherence (either on the basis of a low study-drug concentration or an investigator report), and 10 had an HCV relapse. All 10 patients with a virologic relapse were black, 7 had the TT allele in the gene encoding IL28B (which confers an increased risk of treatment failure with interferon-containing regimens), and 8 received efavirenz (Table S4 in the Supplementary Appendix). To identify which of these characteristics were associated with HCV relapse, exploratory univariate analysis was performed, which identified black race and the presence of the TT allele as significant associations. Although among black patients, relapses occurred in 8 of 61 patients taking efavirenz (13%) and in 2 of 54 patients taking other antiretroviral regimens (4%), the difference was not significant (P=0.10) (Table S5 in the Supplementary Appendix). In the multivariate analysis, black race was the only factor that had an independent association with relapse (odds ratio, 17.73; P=0.001) (Tables S6 and S7 in the Supplementary Appendix). Of the 10 patients who had a relapse, 9 were enrolled in the retreatment substudy at the time of this report.......This association between black race and a decreased rate of virologic response was not observed in the 308 black patients who were monoinfected with HCV receiving ledipasvir-sofosbuvir across the phase 3 program.......The CYP2B6 polymorphism, which is more common among blacks and has been reported to be associated with higher serum efavirenz levels, was assessed in a candidate-gene analysis and was not associated with relapse.26 A genomewide association study might be able to identify genetic factors associated with this observation."

-----------------
Ledipasvir and Sofosbuvir for HCV in Patients Coinfected with HIV-1
NEJM
July 21, 2015
Susanna Naggie, M.D., M.H.S., Curtis Cooper, M.D., Michael Saag, M.D., Kimberly Workowski, M.D., Peter Ruane, M.D., William J. Towner, M.D., Kristen Marks, M.D., Anne Luetkemeyer, M.D., Rachel P. Baden, M.D., Paul E. Sax, M.D., Edward Gane, M.D., Jorge Santana-Bagur, M.D., Luisa M. Stamm, M.D., Ph.D., Jenny C. Yang, Pharm.D., Polina German, Pharm.D., Hadas Dvory-Sobol, Ph.D., Liyun Ni, M.A., Phillip S. Pang, M.D., Ph.D., John G. McHutchison, M.D., Catherine A.M. Stedman, M.B., Ch.B., Ph.D., Javier O. Morales-Ramirez, M.D., Norbert Br�u, M.D., Dushyantha Jayaweera, M.D., Amy E. Colson, M.D., Pablo Tebas, M.D., David K. Wong, M.D., Douglas Dieterich, M.D., and Mark Sulkowski, M.D. for the ION-4 Investigators
Background
Effective treatment for hepatitis C virus (HCV) in patients coinfected with human immunodeficiency virus type 1 (HIV-1) remains an unmet medical need.
Methods
We conducted a multicenter, single-group, open-label study involving patients coinfected with HIV-1 and genotype 1 or 4 HCV receiving an antiretroviral regimen of tenofovir and emtricitabine with efavirenz, rilpivirine, or raltegravir. All patients received ledipasvir, an NS5A inhibitor, and sofosbuvir, a nucleotide polymerase inhibitor, as a single fixed-dose combination for 12 weeks. The primary end point was a sustained virologic response at 12 weeks after the end of therapy.
Results
Of the 335 patients enrolled, 34% were black, 55% had been previously treated for HCV, and 20% had cirrhosis. Overall, 322 patients (96%) had a sustained virologic response at 12 weeks after the end of therapy (95% confidence interval [CI], 93 to 98), including rates of 96% (95% CI, 93 to 98) in patients with HCV genotype 1a, 96% (95% CI, 89 to 99) in those with HCV genotype 1b, and 100% (95% CI, 63 to 100) in those with HCV genotype 4. Rates of sustained virologic response were similar regardless of previous treatment or the presence of cirrhosis. Of the 13 patients who did not have a sustained virologic response, 10 had a relapse after the end of treatment. No patient had confirmed HIV-1 virologic rebound. The most common adverse events were headache (25%), fatigue (21%), and diarrhea (11%). No patient discontinued treatment because of adverse events.
Conclusions
Ledipasvir and sofosbuvir for 12 weeks provided high rates of sustained virologic response in patients coinfected with HIV-1 and HCV genotype 1 or 4. (Funded by Gilead Sciences; ION-4 ClinicalTrials.gov number, NCT02073656.)
Globally, an estimated 4 million to 5 million persons are chronically infected with both human immunodeficiency virus type 1 (HIV-1) and hepatitis C virus (HCV).1 Patients who are coinfected with HIV-1 and HCV have higher rates of cirrhosis, hepatocellular carcinoma, and hepatic decompensation than do patients monoinfected with HCV; they also have a higher rate of death from any cause.2-8 In observational cohort studies, treatment-induced clearance of HCV infection has been associated with decreased morbidity and mortality associated with liver disease.9,10 However, treatment of HCV with interferon and ribavirin in patients who are coinfected with HIV-1 and HCV has historically been associated with low rates of sustained virologic response, high rates of treatment-related cytopenias, and complex interactions with concomitant antiretroviral drugs.11-13 The first oral HCV direct-acting antiviral drugs - the NS3/4A protease inhibitors boceprevir and telaprevir - were not approved by the Food and Drug Administration for patients coinfected with HIV-1 and HCV.14,15
The phase 3 PHOTON-1 and PHOTON-2 studies investigated the safety and efficacy of the nucleotide NS5B polymerase inhibitor sofosbuvir in combination with ribavirin for the treatment of HCV in patients coinfected with HIV-1.16,17 Outcomes from these studies compared favorably to those reported for the protease inhibitor-containing regimens. However, oral regimens of direct-acting antiviral drugs that combine more than one potent antiviral agent appear to offer improved rates of response over those seen with a single direct-acting antiviral drug plus ribavirin with or without peginterferon.18,19 One such regimen, the fixed-dose combination of sofosbuvir and ledipasvir (an inhibitor of nonstructural protein 5A [NS5A], which has an important role in HCV RNA replication), was recently approved for the treatment of chronic genotype 1 HCV in the United States and genotype 1 and 4 HCV in the European Union. In a phase 2 study evaluating 12 weeks of ledipasvir-sofosbuvir in a cohort of mostly black patients (84%) coinfected with HCV genotype 1 and HIV-1, the rate of sustained virologic response was 98%.20 We conducted a larger phase 3 trial, called the ION-4 study, to evaluate 12 weeks of treatment with ledipasvir-sofosbuvir in patients with HIV-1 who were coinfected with HCV genotype 1 or 4, including patients with compensated cirrhosis and those in whom previous treatment with an HCV regimen containing peginterferon, an HCV protease inhibitor, or direct-acting antiviral drugs including sofosbuvir had failed.21
Methods
Patients
From March 7, 2014, to June 9, 2014, we enrolled patients who were 18 years of age or older at 60 sites in the United States, Puerto Rico, Canada, and New Zealand. Patients were required to be receiving a stable, protocol-approved antiretroviral regimen for HIV-1 for at least 8 weeks before screening and to have evidence of HIV-1 viral suppression (HIV-1 RNA, <50 copies per milliliter) with a CD4+ count of more than 100 cells per microliter. On the basis of drug-interaction data in healthy volunteers that were available at the time of protocol development, allowable antiretroviral drugs included emtricitabine and tenofovir disoproxil fumarate plus efavirenz, raltegravir, or rilpivirine.22
A minimum creatinine clearance of 60 ml per minute, as calculated by the Cockcroft-Gault equation, was required for enrollment. Planned enrollment included approximately 50% of patients who had previously been treated for HCV (and in whom an oral regimen of sofosbuvir plus ribavirin had failed in 13 patients) and 20% with compensated cirrhosis. Cirrhosis was defined as histopathological evidence of cirrhosis as follows: stage 4 fibrosis on the Metavir scale, which ranges from 0 to 4, with higher stages indicating a greater degree of fibrosis; or a score of 5 or 6 on the Ishak fibrosis scale, which ranges from 0 to 6, with higher scores indicating more extensive fibrosis and scores of 5 or higher indicating cirrhosis. In addition, all patients were required to have a score of more than 12.5 kPa on transient elastography testing or a FibroTest score of more than 0.75 together with a ratio of aspartate aminotransferase to platelets of more than 2. Patients with a history of alcohol or drug abuse within 12 months before screening were not eligible. Race was self-reported. Full eligibility criteria are provided in the study protocol, available with the full text of this article at NEJM.org.
Study Design
In this multicenter, open-label trial, all patients received a fixed-dose combination tablet containing 90 mg of ledipasvir and 400 mg of sofosbuvir, administered orally once daily for 12 weeks. Patients who had a virologic relapse after completing therapy were eligible for retreatment with ledipasvir-sofosbuvir plus ribavirin for 24 weeks.
Study Assessments
Screening assessments included serum HCV RNA levels, HIV RNA levels, and IL28B (rs12979860) genotyping, as well as standard laboratory and clinical testing. Serum HCV RNA was measured with the COBAS TaqMan HCV Test (version 2.0) for use with the High Pure System (HPS, Roche Molecular Systems), which has a lower limit of quantification of 25 IU per milliliter. HCV genotype and subtype were determined with the use of the Versant HCV Genotype INNO-LiPA 2.0 assay (Siemens). HIV RNA was measured by means of the AmpliPrep/COBAS TaqMan HIV-1 Test, version 2.0, which has a lower limit of quantification of 25 copies per milliliter.
Assessments during treatment included standard laboratory testing and measurements of plasma HCV RNA and HIV-1 RNA levels, along with evaluations of adherence, measurement of vital signs, electrocardiography, and symptom-directed physical examinations. All adverse events were recorded and graded according to a standardized scale. (Details are provided in the study protocol.) Patients with plasma HIV-1 RNA levels of 400 copies per milliliter or higher at two or more consecutive post-baseline visits at least 2 weeks apart were considered to have HIV-1 virologic rebound.
Modestly increased levels of tenofovir (by a factor of 1.3 to 1.8, as compared with levels in patients receiving antiretroviral drugs alone) were observed in studies involving healthy volunteers in whom non-nucleoside reverse transcriptase-based regimens containing tenofovir disoproxil fumarate were administered with ledipasvir-sofosbuvir.22 For this reason, monitoring of renal function was performed in all patients. (See the Supplementary Appendix, available at NEJM.org, for details regarding renal monitoring.)
Samples for pharmacokinetic analyses were collected from all patients at baseline and at weeks 1, 2, 4, 6, 8, 10, and 12 (including at early termination visits). All patients were also eligible to participate in an optional pharmacokinetic substudy to determine the steady-state pharmacokinetics of ledipasvir, sofosbuvir, GS-331007 (the predominant circulating metabolite of sofosbuvir), and tenofovir. (See the Supplementary Appendix for details.)
For analysis of HCV viral resistance, deep sequencing of the NS5A and nonstructural protein 5B (NS5B) regions of the HCV RNA was performed at baseline in all patients. For patients with virologic failure, deep sequencing was performed with samples collected at the time of the first virologic failure. Variants that were present in at least 1% of the viral population were reported.
Study Oversight
The trial was approved by the institutional review board or independent ethics committee at each participating site and was conducted in compliance with the provisions of the Declaration of Helsinki, Good Clinical Practice guidelines, and local regulatory requirements. The sponsor (Gilead) collected the data, monitored study conduct, and performed the statistical analyses. An independent data and safety monitoring committee reviewed the progress of the study. All the authors vouch for the completeness and accuracy of the data and data analyses and for the fidelity of this report to the study protocol. The first author wrote the first draft of the manuscript. All authors reviewed the manuscript and provided input. Editorial assistance was provided by an employee of Gilead Sciences.
Study End Points
The primary efficacy end point was the rate of sustained virologic response, which was defined as the absence of quantifiable HCV RNA in serum (<25 IU per milliliter) at 12 weeks after the end of therapy. Sustained virologic response 24 weeks after the end of treatment was a secondary end point.
The primary safety end point was any adverse event leading to permanent discontinuation of study treatment. A secondary safety end point was the proportion of patients who maintained HIV-1 viral suppression (HIV-1 RNA, <50 copies per milliliter) while receiving HCV treatment. Exploratory prespecified subgroup analyses were performed to examine the association between baseline characteristics and the primary efficacy end point.
Statistical Analysis
We calculated the proportion of patients who had a sustained virologic response along with exact two-sided 95% confidence intervals using the Clopper-Pearson method. With approximately 300 patients, the two-sided 95% confidence interval for the primary end point was expected to extend no more than 3.4% in both directions from the observed rate on the assumption that the response rate would be 90%. An exploratory exact logistic-regression analysis was performed to identify baseline factors that were independently associated with relapse. (See the Supplementary Appendix for methods used in the regression analysis.)
Results
Study Patients
A total of 429 patients were screened for enrollment (Table S1 and Fig. S1 in the Supplementary Appendix). Of these patients, 335 were enrolled and began treatment. Seventy-five percent of patients were infected with HCV genotype 1a, 23% with HCV genotype 1b, and 2% with HCV genotype 4 (Table 1). Overall, 34% of patients were black, 82% were male, 20% had compensated cirrhosis, and 55% had received previous unsuccessful treatment for HCV (of whom 36% had received previous direct-acting antiviral drugs) (Table S2 in the Supplementary Appendix). Among the 67 patients with cirrhosis, the median baseline albumin level was 3.7 g per deciliter, the median platelet count was 137,000 per microliter, and the median international normalized ratio (INR) was 1.1. The median CD4+ count at baseline was 628 cells per microliter; the CD4+ count was under 200 cells per microliter in 4 patients and under 350 cells per microliter in 37 patients. All patients were receiving emtricitabine and tenofovir disoproxil fumarate, along with efavirenz (in 48%), raltegravir (in 44%), or rilpivirine (in 9%).
Efficacy
Among the 335 patients who were enrolled and treated, 322 (96%; 95% confidence interval [CI], 93 to 98) had a sustained virologic response 12 weeks after the end of therapy (Table 2). Of the 322 patients with a response, 312 returned for the post-treatment week 24 visit, at which all the patients had a sustained virologic response.
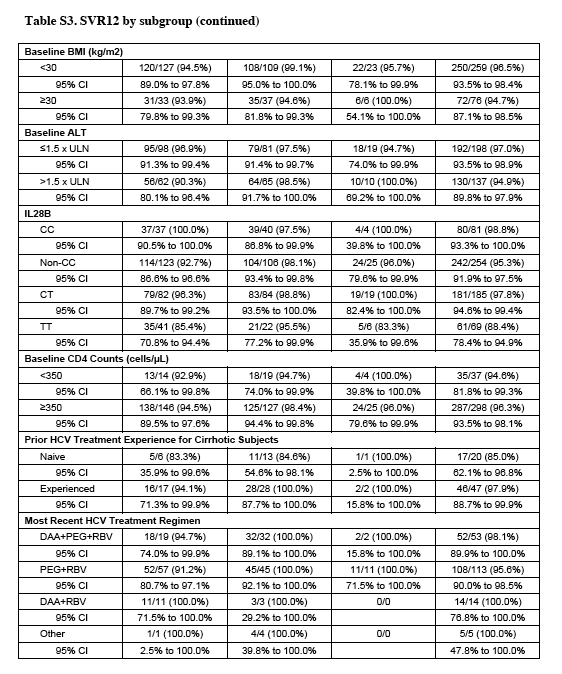
The rates of response at 12 weeks were similar in patients with genotype 1a and those with 1b, in men and women, in patients who had undergone previous treatment and those who had not, in patients receiving various concomitant HIV antiretroviral regimens, and in patients with cirrhosis (including those who had received previous treatment) and those without cirrhosis (Table S3 and Fig. S2 in the Supplementary Appendix). Black patients had lower response rates than did patients of other races (90% [95% CI, 83 to 95] vs. 99% [95% CI, 97 to 100], P<0.001 by Fisher's exact test). All 13 patients who had a relapse after completing 12 or 24 weeks of previous treatment with sofosbuvir plus ribavirin had a sustained virologic response.
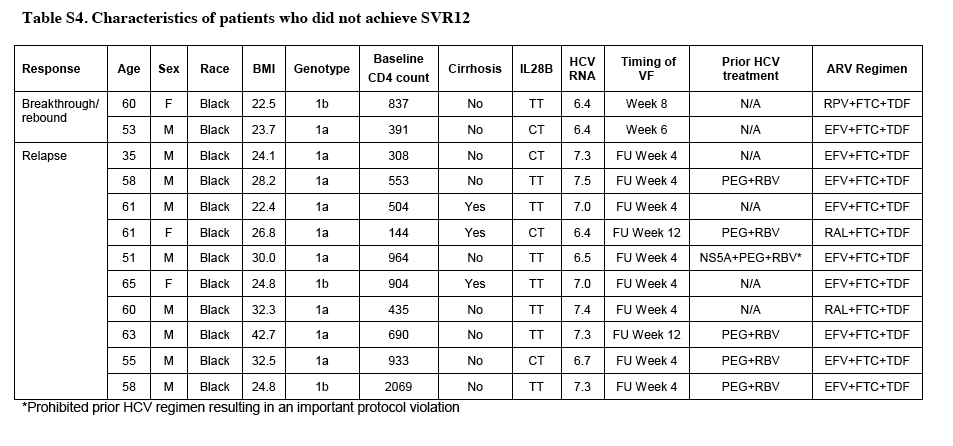
In total, 13 patients (4%) did not have a sustained virologic response. Of these patients, 1 died after 4 weeks of treatment, 2 had HCV breakthrough during treatment that was associated with suspected poor adherence (either on the basis of a low study-drug concentration or an investigator report), and 10 had an HCV relapse. All 10 patients with a virologic relapse were black, 7 had the TT allele in the gene encoding IL28B (which confers an increased risk of treatment failure with interferon-containing regimens), and 8 received efavirenz (Table S4 in the Supplementary Appendix). To identify which of these characteristics were associated with HCV relapse, exploratory univariate analysis was performed, which identified black race and the presence of the TT allele as significant associations. Although among black patients, relapses occurred in 8 of 61 patients taking efavirenz (13%) and in 2 of 54 patients taking other antiretroviral regimens (4%), the difference was not significant (P=0.10) (Table S5 in the Supplementary Appendix). In the multivariate analysis, black race was the only factor that had an independent association with relapse (odds ratio, 17.73; P=0.001) (Tables S6 and S7 in the Supplementary Appendix). Of the 10 patients who had a relapse, 9 were enrolled in the retreatment substudy at the time of this report.
Virologic Resistance Testing
Before undergoing treatment, 59 of 325 patients (18%) with genotype 1 HCV infection were found to have resistance-associated NS5A variants that confer reduced susceptibility to ledipasvir. Of these 59 patients, 55 (93%) had a sustained virologic response at 12 weeks, whereas 258 of 266 patients (97%) who did not have resistance-associated NS5A variants at baseline had such a response (P=0.24 by Fisher's exact test). The 2 patients who had on-treatment virologic failure did not have NS5A variants at baseline but did have such emergent variants at the time of treatment failure. Of the 10 patients with virologic relapse, NS5A variants were detected in 4 patients at baseline and in 8 patients at the time of relapse. The resistance-associated NS5B variant S282T was not detected in any patient at baseline or at the time of virologic failure. Of the 40 patients who had received previous treatment with sofosbuvir, 4 (10%) had the treatment-emergent NS5B variant L159F at baseline; these 4 patients had a sustained virologic response to ledipasvir-sofosbuvir at 12 weeks. Of the 10 patients with HCV relapse, 1 had both L159F and resistance-associated NS5A variants at the time of relapse.
Safety
None of the 335 patients in the study discontinued treatment prematurely because of an adverse event. Overall, 257 patients (77%) had an adverse event, most of which were mild to moderate in severity (Table 3). Eight patients had 15 serious adverse events. The only serious adverse events that occurred in more than 1 patient were hepatocellular carcinoma (in 2 patients) and portal-vein thrombosis (in 2); all events were reported in patients with cirrhosis. Three patients had serious infections: sepsis during the fifth week of the study, spontaneous bacterial peritonitis during the fourth week of follow-up, and serious respiratory infection at the end of the second week of follow-up and Clostridium difficile colitis during the sixth week of follow-up. (A full list of serious adverse events is provided in Table S8 in the Supplementary Appendix.)
One patient died after discontinuing treatment early. This patient, a 59-year-old white man with confirmed intravenous drug use, received the diagnosis of Staphylococcus aureus endocarditis and sepsis on day 41 of treatment.
There were reports of laboratory abnormalities of grade 3 in 30 patients (9%) and grade 4 in 6 patients (2%) (Table S9 in the Supplementary Appendix). Grades 3 and 4 serum laboratory abnormalities that were reported in more than 1% of patients included elevations in lipase, creatine kinase, and serum glucose. No patient had a clinical episode of pancreatitis. Five patients (1%) had isolated grade 3 or 4 elevations in creatine kinase levels; all were confirmed by the investigator to be in the context of exercise or illicit drug use associated with rhabdomyolysis. Hyperglycemia was reported in 5 patients (1%), all of whom had known diabetes or an abnormal glycated hemoglobin level at baseline. CD4+ counts were stable during treatment, and no patient had HIV-1 virologic failure.
No patient had grade 3 or 4 elevations in serum creatinine, bicarbonate, or potassium or in urinary protein. There was no significant change in urinary levels of ß2-microglobulin or retinol-binding protein, as compared with serum creatinine levels, during the study period. Grade 3 hypophosphatemia was reported in one patient during a single visit and resolved on repeat testing. Four patients had confirmed increases of 0.4 mg per deciliter (35 μmol per liter) or more in serum creatinine levels; one discontinued tenofovir, and one had a dose reduction of tenofovir; the other two completed treatment with no alteration in the antiretroviral regimen. (Details regarding these patients are provided in the Supplementary Appendix.)
Pharmacokinetics
There were no clinically relevant differences in the levels of sofosbuvir, GS-331007 (sofosbuvir metabolite), or ledipasvir in subgroups of patients (black vs. nonblack, those with a virologic response vs. those with virologic failure, and those receiving an efavirenz-containing regimen vs. those receiving other regimens). There was no significant difference in the mean plasma area under the curve for tenofovir on the basis of either the antiretroviral regimen or the change in serum creatinine from baseline (Table S10 in the Supplementary Appendix).
Discussion
In this multicenter, open-label, single-group study, 12 weeks of treatment with the once-daily, single-tablet regimen of ledipasvir-sofosbuvir resulted in a sustained virologic response in 96% of patients. In exploratory subgroup analyses, rates of sustained virologic response 12 weeks after the end of therapy (the primary efficacy end point) were similar across all subgroups except that black patients, who made up 34% of the study population, had lower rates of sustained virologic response. This association between black race and a decreased rate of virologic response was not observed in the 308 black patients who were monoinfected with HCV receiving ledipasvir-sofosbuvir across the phase 3 program.23-25 The CYP2B6 polymorphism, which is more common among blacks and has been reported to be associated with higher serum efavirenz levels, was assessed in a candidate-gene analysis and was not associated with relapse.26 A genomewide association study might be able to identify genetic factors associated with this observation.
Ledipasvir-sofosbuvir has limited potential for clinically significant drug interactions with most antiretroviral agents.19,22 However, results from phase 1 evaluations showed that concomitant administration of ledipasvir-sofosbuvir and tenofovir disoproxil fumarate as a component of an antiretroviral regimen resulted in modest increases (approximately 40%) in the exposure to tenofovir, as compared with an antiretroviral regimen alone.19 In accordance with these findings, administration of emtricitabine plus tenofovir disoproxil fumarate with ledipasvir-sofosbuvir in patients coinfected with HCV and HIV-1 resulted in moderately higher tenofovir exposures than those reported with antiretroviral regimens alone, including those involving either a non-nucleoside or non-nucleotide reverse-transcriptase inhibitor or an integrase strand-transfer inhibitor. Intensive renal monitoring, including evaluation of urine biomarkers, revealed that four patients had treatment-emergent worsening of renal function.
Limitations of this study include its single-group, open-label design and the restriction of permitted antiretroviral regimens. Open-label studies are at risk for bias in areas that include the selection and retention of patients and outcome reporting. Single-group studies cannot adequately control for confounding, and multiple-subgroup analyses are at risk for type I error. Furthermore, patients taking ritonavir-boosted HIV-1 protease inhibitors or cobicistat-boosted elvitegravir with tenofovir disoproxil fumarate were excluded from the study owing to the potential for additional increases in tenofovir exposure. The results from a recent phase 1 study that evaluated drug interactions between ritonavir-boosted HIV-1 protease inhibitors with emtricitabine-tenofovir disoproxil fumarate and ledipasvir-sofosbuvir confirmed a relative increase of 30 to 60% in the exposure to tenofovir, as compared with antiretroviral therapy alone.27 Thus, the safety of this HCV combination in patients with HIV-1 infection who are receiving these antiretroviral regimens is unknown.
In conclusion, we found that a fixed-dose combination of ledipasvir plus sofosbuvir for 12 weeks provided high rates of sustained virologic response in patients with HCV genotype 1 or 4 who were coinfected with HIV-1, including those who had previous treatment failure while receiving regimens that included direct-acting antiviral drugs and those with cirrhosis. Response rates in the study were similar to those seen in the phase 3 registration trials for this regimen in HCV-monoinfected patients.
|
| |
|
|
|
|
|