| |
EASL Recommendations on Treatment of Hepatitis C 2016
|
| |
| |
Download the PDF here
European Association for the Study of the Liver
published Jan 2017 Jnl of Hepatology
from Jules: attached is full pdf.
Below are excerpts.
Key Topics
treatments available, recommended;
PWIDs; drug-drug interactions,
ART drug-drug interactions;
post SVR followup for patients, with cirrhosis;
resistance testing;
retreatment options for those not achieving SVR with prior treatment; acute HCV treatment.
Post-treatment follow-up of patients who achieve an SVR
Non-cirrhotic patients who achieve an SVR should be retested for HCV RNA (or HCV core antigen if HCV RNA testing is not available or not affordable) at 48 weeks post-treatment. If HCV RNA (or HCV core antigen) is still not detected, the infection can be considered as definitely cured and HCV RNA does not need to be retested. Patients with pre-existing cofactors for liver disease (notably, history of alcohol drinking, obesity and/or type 2 diabetes) should be carefully and periodically subjected to a thorough clinical assessment, as needed.
Cirrhotic patients who achieve an SVR should remain under surveillance for HCC every 6 months by ultrasound (like patients with advanced fibrosis, METAVIR score F3), and for oesophageal varices by endoscopy if varices were present at pre-treatment endoscopy (though first variceal bleed is seldom observed after SVR). The presence of cofactors for liver disease, such as history of alcohol drinking, obesity and/or type 2 diabetes, may determine that additional assessments are necessary. Long-term post-SVR follow-up studies showed that, although it is significantly reduced compared to untreated patients or patients who did not achieve an SVR, the risk of developing HCC remains in patients with cirrhosis who eliminate HCV [[3], [4]]. Thus, the duration of HCC surveillance in patients with advanced fibrosis or cirrhosis who achieve an SVR is indefinite.
There remains some concern that reinfection due to recurrent or persistent risk behaviour may negate the potential benefit of treatment. Reported rates of reinfection following successful HCV treatment among patients at high-risk, such as PWID or men who have sex with men, are in the order of 1-8% per year [[165], [166], [167], [168], [169]]. The ease of IFN-free therapy may increase the likelihood of reinfection, as recently suggested [170]. In order to maximize the benefit of therapy, the risks of reinfection should be emphasized to patients at risk, and behavioural modifications should be positively reinforced.
HCV resistance testing
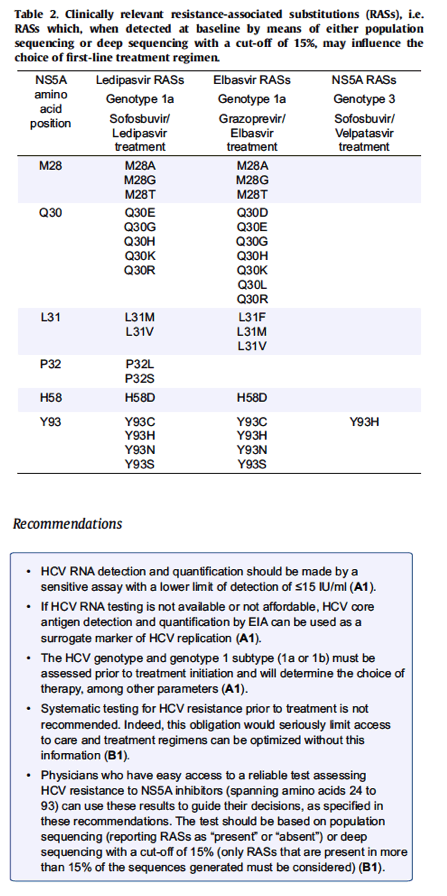
No standardized tests for the resistance of HCV to approved drugs are available as purchasable kits. Resistance testing relies on in-house techniques based on population sequencing (Sanger sequencing) or deep sequencing [29]. A limited number of laboratories have made such tests available in Europe and in other continents. HCV resistance testing may be technically difficult, in particular for genotypes other than 1 and 4, and the performances of the available in-house assays widely vary. Thus, access to HCV resistance testing remains limited.
Because access to reliable HCV resistance testing is limited and there is no consensus on the techniques or the interpretation and the reporting of these tests, systematic testing for HCV resistance prior to treatment is not recommended [30]. Indeed, systematic testing would seriously limit access to care, whereas treatment can be optimized for groups of patients with the risk that the presence of resistance-associated substitutions (RASs) at baseline reduces response to therapy.
Physicians who have easy access to reliable resistance tests can use these results to guide their decisions. Only the NS5A region, the target of NS5A inhibitors, should be analysed. The test should be based on population sequencing (reporting RASs as "present" or "absent") or deep sequencing with a cut-off of 15% (only RASs that are present in more than 15% of the sequences generated are clinically significant and should be considered). The test should be able to reliably determine the sequence of a region spanning NS5A amino acids 24 to 93. The genotype-specificity of the test should be specified. Table 2 presents RASs that are clinically relevant, i.e. the presence of which may influence decision on the treatment regimen if the resistance test is performed.
Indications for treatment: who should be treated?
To succeed, HCV elimination will require national plans together with forecasted budgeting to expedite unrestricted access to treatment.
All treatment-naïve and treatment-experienced patients with compensated or decompensated chronic liver disease related to HCV, who are willing to be treated and who have no contraindications to treatment, must be considered for therapy.
Treatment must be considered without delay in patients with significant fibrosis (METAVIR score F2 or F3) or cirrhosis (METAVIR score F4), including decompensated cirrhosis; patients with clinically significant extrahepatic manifestations (e.g. symptomatic vasculitis associated with HCV-related mixed cryoglobulinaemia, HCV immune complex-related nephropathy and non-Hodgkin B cell lymphoma); patients with HCV recurrence after liver transplantation; patients at risk of a rapid evolution of liver disease due to concurrent comorbidities (non-liver solid organ or stem cell transplant recipients, diabetes); and individuals at risk of transmitting HCV (active injection drug users, men who have sex with men with high-risk sexual practices, women of childbearing age who wish to get pregnant, haemodialysis patients, incarcerated individuals). Injection drug users and men who have sex with men with high-risk sexual practices should be made aware of the risk of reinfection and should apply preventive measures after successful treatment. Patients with decompensated cirrhosis and an indication for liver transplantation with a MELD score ⩾18-20 will benefit from transplantation first and antiviral treatment after transplantation, because the probability of significant liver function improvement and delisting is low. However, patients with a MELD score ⩾18-20 with a waiting time before transplantation expected to be more than six months can be treated for their HCV infection.
Treatment is not recommended in patients with limited life expectancy due to non-liver-related comorbidities.

Active drug addicts and patients on stable maintenance substitution
Ageing cohorts of people who inject drugs (PWID) with chronic HCV and low treatment uptake are making a significant contribution to the population with advanced liver disease and to liver-related mortality [[131], [132]]. The prevalence of HCV among PWIDs is approximately 65% [[133], [134], [135]] and >80% among long-term PWIDs [133].
HCV treatment must be considered for PWIDs who are willing to receive treatment, are able and willing to maintain regular appointments and adherence, and accept to undergo integrated management of their substance abuse, including syringe exchange program, substitution therapy and other general harm reduction strategies. Guidelines for pre-therapeutic assessment and care of HCV infected PWIDs are available [[38], [136]]. Modelling studies suggest that implementation of HCV treatment for PWIDs could reduce transmission when combined with preventive measures [[137], [138], [139]]. Composite screening, linkage to care and treatment, together with harm reduction programs, are urgently required for this important reservoir. Decisions to treat must be made on a case-by-case basis. PWIDs with on-going social issues and/or with a history of psychiatric disease or with more frequent drug use during therapy are at risk of lower adherence and reduced likelihood of achieving SVR and need to be monitored closely during therapy, and also need more supporting measures.
HCV treatment has been delivered successfully to drug users through various clinical models, including within general hospital liver disease and viral hepatitis clinics, drug detoxification clinics, opioid substitution therapy clinics, prisons and community-based clinics.
DAA clinical development programs have excluded individuals with active drug use, but many trials have included those on opioid substitution therapy. DAA-based safety and treatment outcome data have not been presented on clinical trial sub-populations of individuals on opioid substitution therapy. In a Phase II study, patients infected with HCV genotype 1 without cirrhosis who were treatment-naïve and on chronic methadone or buprenorphine substitution were treated with the combination of ritonavir-boosted paritaprevir, ombitasvir and dasabuvir for 12 weeks with ribavirin. The SVR rate was 97% (37/38) [140]. In the C-EDGE CO-STAR trial, treatment-naïve patients on opioid substitution for at least 3 months were treated with grazoprevir and elbasvir for 12 weeks without ribavirin. The SVR12 rates were 93% (144/154) in genotype 1a patients, 93% (28/30) in genotype 1b patients, and 92% (11/12) in genotype 4 patients. Study medication adherence was high and safety was not different from that in the placebo arm [141]. Studies are on-going with other treatment regimens in PWIDs on opioid substitution.
Some drug-drug interaction studies have been undertaken between individual DAAs and either methadone or buprenorphine. The only potentially clinically important interaction observed is between ritonavir-boosted paritaprevir and ombitasvir plus dasabuvir with buprenorphine, with a recommendation to monitor closely. However, given that actual pharmacokinetic interaction data are not available for all the DAA regimens with both methadone and buprenorphine, it is prudent to monitor for signs of opioid toxicity.
In addition to opioid substitution therapy, antidepressants, antipsychotics and sedatives are frequently used by patients with addiction problems. There is a lack of formal pharmacokinetic studies between many of the psychoactive drugs and DAAs. Escitalopram and citalopram have been studied and either of these drugs can be safely combined with HCV treatment [142]. Ritonavir-boosted paritaprevir and ombitasvir with dasabuvir are most likely to cause drug interactions via the inhibition of CYP3A4. Caution is thus warranted when drugs with a narrow therapeutic index, such as midazolam and quetiapine, are co-administered with these DAAs. Pharmacokinetic studies on recreational and illicit drug use have not been performed.
Reinfection rates will require reporting and monitoring, and appropriate interventions to limit retreatment will be necessary.

Available drugs in Europe in 2016
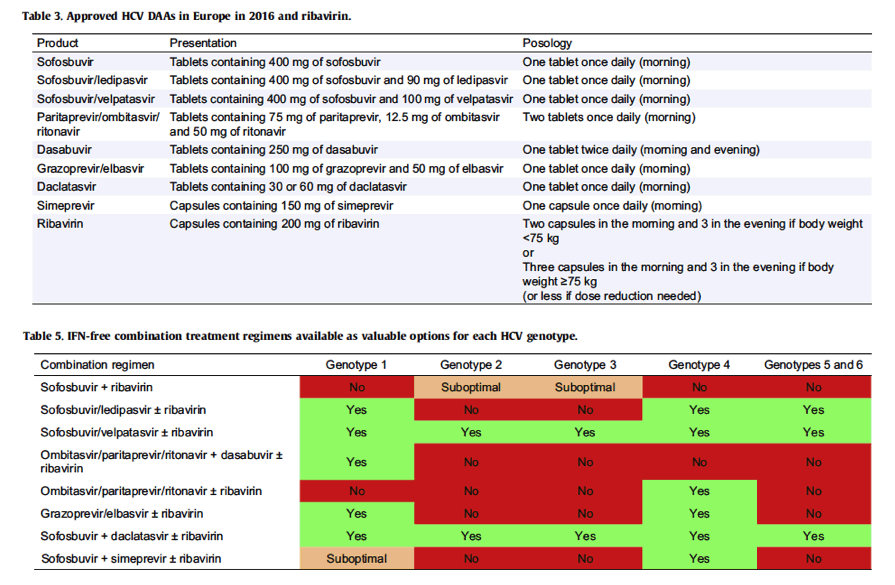
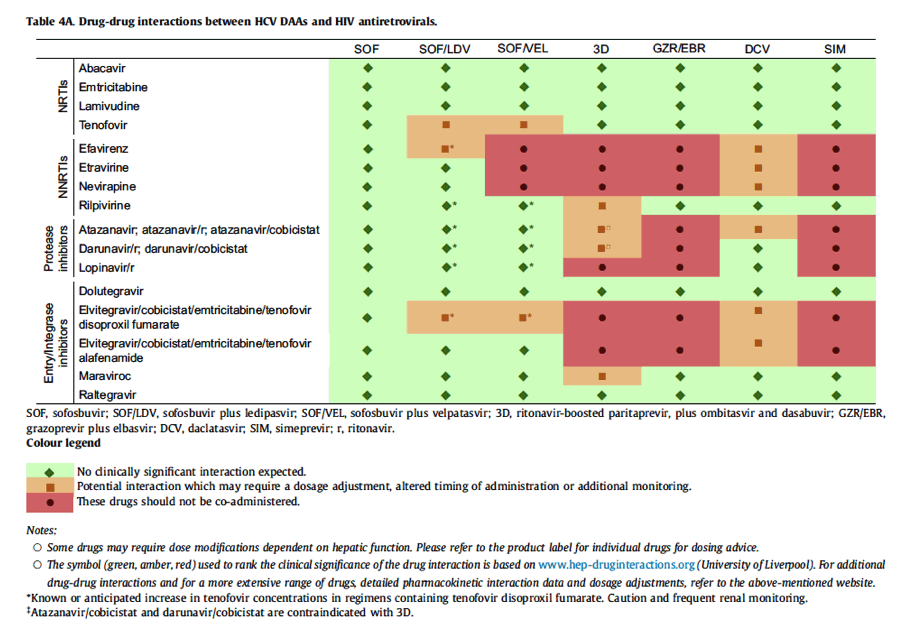
The HCV drugs available in Europe are listed in this paragraph and in Table 3. Their known pharmacokinetic profiles and how this impacts drug-drug interactions are presented. For a more comprehensive listing of drug-drug interactions, see Table 4A, Table 4B, Table 4C, Table 4D, Table 4E, Table 4F and www.hep-druginteractions.org. For additional information on the disposition of individual DAAs, refer to the Summary of Product Characteristics.
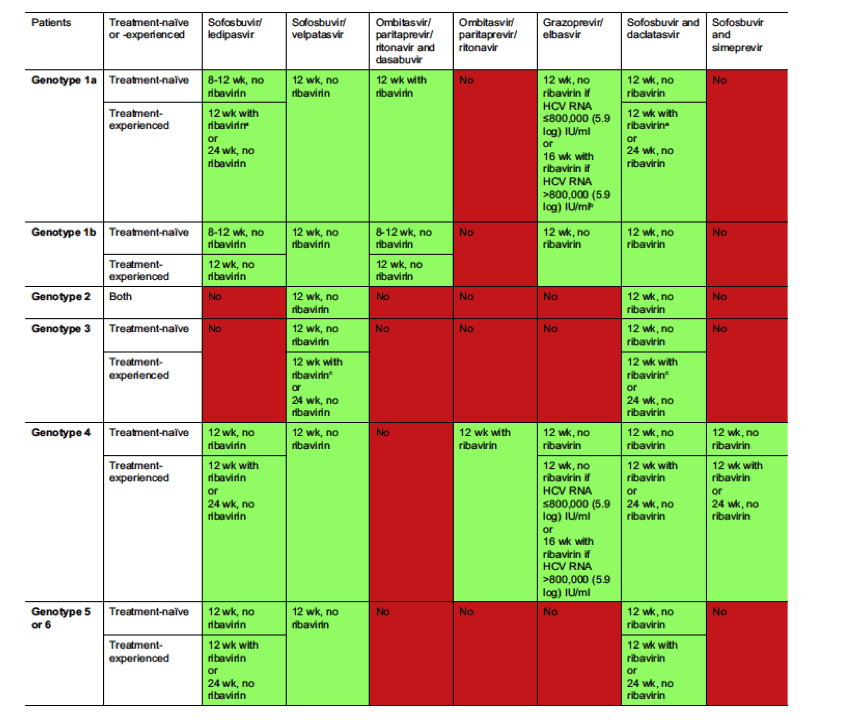
Treatment of HCV genotype 1 infection
Five treatment options are available in 2016 for patients infected with HCV genotype 1 (Table 5). The combination of sofosbuvir and simeprevir was shown to yield lower SVR12 rates than other combinations of DAAs in real-world studies and is therefore not recommended as an option equivalent to the others [[33], [34], [35], [36]]. However, in areas where it is the only available IFN-free option, the combination of sofosbuvir and simeprevir with or without ribavirin can be used to treat genotype 1 infection, according to prior recommendations [37].
In settings where none of the proposed IFN-free options is available, the double combination of pegylated IFN-α and ribavirin, or the triple combination of pegylated IFN-α, ribavirin and telaprevir, boceprevir, simeprevir or sofosbuvir remain acceptable for patients likely to respond to these regimens until new DAAs become available and affordable; see prior EASL Clinical Practice Guidelines [[37], [38], [39]].


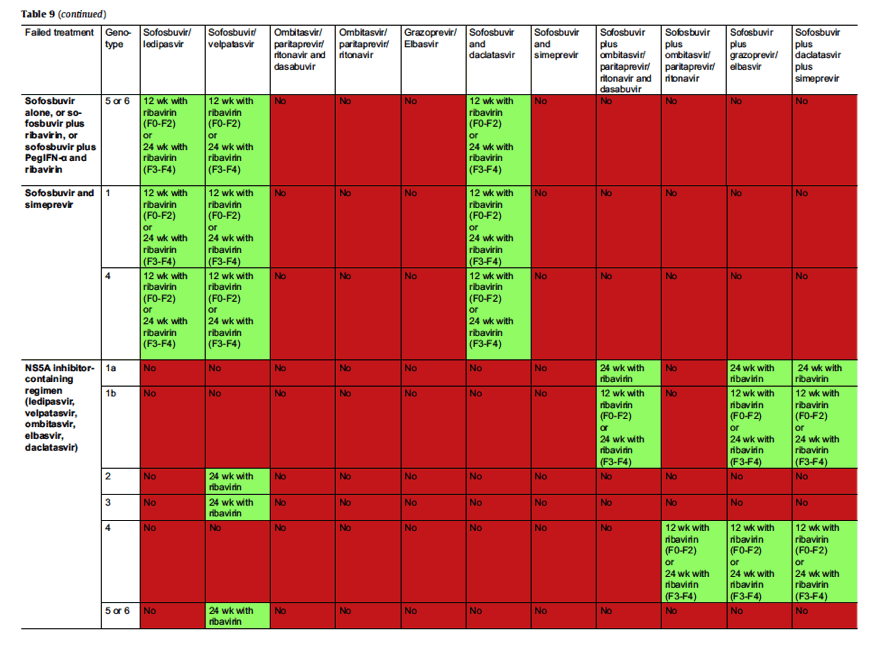
Retreatment of patients who failed after an IFN-free regimen (all genotypes)
Sofosbuvir has a high barrier to resistance. Clinically meaningful resistant HCV variants have been exceptionally reported with sofosbuvir, and they rapidly disappeared after treatment cessation [[30], [50]]. Thus, retreatment strategies should include sofosbuvir. In contrast, patients exposed to a protease inhibitor (paritaprevir, grazoprevir, simeprevir), an NS5A inhibitor (ledipasvir, velpatasvir, ombitasvir, elbasvir, daclatasvir) or a non-nucleoside inhibitor of HCV polymerase (dasabuvir) who fail to achieve SVR select viruses with RASs in the NS3 protease, NS5A and polymerase regions, respectively. Viruses resistant to protease inhibitors progressively decrease in proportion to become undetectable by means of population sequencing (direct sequence analysis) within a few months to 2 years after treatment cessation. In contrast, viruses resistant to NS5A inhibitors are fit and remain dominant for many years, perhaps forever, after they have been selected by a regimen including an NS5A inhibitor [[30], [50]].
Patients who failed on a DAA-containing regimen should be retreated with an IFN-free combination including a drug with a high barrier to resistance (currently, sofosbuvir), plus one to three other drugs, ideally with no cross-resistance with the drugs already administered. Retreatment should be for 12 weeks with ribavirin, or extended to 24 weeks with ribavirin in more difficult-to-cure patients, such as patients with F3 fibrosis or cirrhosis, or 24 weeks without ribavirin for those who have a contraindication or do not tolerate ribavirin.
Currently, only a few studies including a small number of selected patients support these retreatment recommendations, which are mostly based on indirect evidence. In a study, retreatment with 12 weeks of sofosbuvir plus ledipasvir with ribavirin yielded SVR in 98% (50/51) of genotype 1 patients who failed prior treatment with sofosbuvir plus placebo, or sofosbuvir plus ribavirin, or sofosbuvir plus pegylated IFN-α and ribavirin [171]. In another study, 15 patients who failed to achieve SVR after treatment containing an NS5A inhibitor were retreated with sofosbuvir and simeprevir for 12 weeks without ribavirin. SVR12 was achieved in 8/10 patients with genotype 1a, 3/3 patients with genotype 1b, and 2/2 patients with genotype 4 [172]. Ten patients with F3 fibrosis or compensated cirrhosis who failed to achieve SVR after an IFN-free regimen were retreated with the triple combination of sofosbuvir, simeprevir and daclatasvir with ribavirin for 24 weeks. Six of them achieved SVR12, two relapsed after retreatment, and two patients experienced severe adverse events leading to treatment discontinuation, including one patient who died from acute-on-chronic liver failure [173].
Patients treated with the fixed-dose combination of sofosbuvir and velpatasvir for 4 to 12 weeks who failed to achieve SVR were retreated with the same combination for 24 weeks with ribavirin. Among them, SVR12 was achieved in 97% (33/34) of patients infected with genotype 1, 91% (13/14) of patients infected with genotype 2 and 76% (13/17) of patients infected with genotype 3 [174].
In the QUARTZ-1 study, 20 patients infected with genotype 1 with a history of previous DAA treatment failure without discontinuation for reasons other than virological failure were treated with a combination of sofosbuvir, ritonavir-boosted paritaprevir, ombitasvir and dasabuvir for 12 or 24 weeks, with or without ribavirin. SVR12 was achieved in 13/14 patients with genotype 1a infection without cirrhosis treated for 12 weeks with ribavirin, in 6/6 patients infected with genotype 1a with cirrhosis treated for 24 weeks with ribavirin, and in 2/2 patients infected with genotype 1b treated for 12 weeks without ribavirin [175]. In C-SWIFT-RETREATMENT, patients infected with genotype 1 exposed to a short treatment (4, 6 or 8 weeks) with the combination of sofosbuvir, grazoprevir and elbasvir without ribavirin were retreated with the same drug combination with ribavirin for 12 weeks. All of them (23/23) achieved SVR.
Whether HCV resistance testing prior to retreatment is helpful to make a decision remains unknown, as well as which therapeutic decision should be made based on this result. Table 8 summarizes the RASs that have been shown to confer reduced susceptibility to the corresponding drug class in vitro and/or that have been reported to be selected by IFN-free therapies in patients who failed to achieve SVR. These many RASs and a number of alternative substitutions at the same positions can be present at retreatment baseline in patients previously exposed to DAAs. In the current state of knowledge, no specific algorithms can be derived from these observations to guide retreatment decisions. Thus, retreatment must be guided either by the knowledge of which drugs were administered in previous treatment courses if no resistance test is available or, if resistance testing is performed, by probabilities of response according to the resistance profile observed and the treating team's experience. Preliminary data in a small number of patients suggests that retreatment can be optimized based on RAS testing [176].
---------------
In HIV-HCV coinfected patients, sofosbuvir/velpatasvir may be given with most antiretrovirals, the exceptions being the inducing drugs efavirenz, etravirine and nevirapine. Efavirenz causes a 50% decrease in velpatasvir exposure.
Sofosbuvir/velpatasvir also increases tenofovir exposure due to P-gp inhibition. This means that patients on a regimen containing tenofovir disoproxil fumarate will need to be monitored for renal adverse events.
Sofosbuvir/ledipasvir may be given with all antiretrovirals. However, due to an increase in tenofovir concentrations when a pharmacokinetic enhancer (ritonavir or cobicistat) is present in an antiretroviral regimen, these combinations (i.e. atazanavir/ritonavir, darunavir/ritonavir, lopinavir/ritonavir, elvitegravir/cobicistat, atazanavir/cobicistat, darunavir/cobicistat, all in combination with tenofovir disoproxil fumarate/emtricitabine) should be used with caution, with frequent renal monitoring if other alternatives are not available. The interaction is not mitigated by staggering administration by 12 h. Tenofovir is also increased in efavirenz-containing regimens and caution is required. The recent approval of tenofovir alafenamide (TAF), giving much reduced plasma tenofovir exposure, means that there is less concern about an interaction leading to increased tenofovir exposure.
Like ledipasvir, the solubility of velpatasvir decreases as pH increases. Therefore, it is important to be aware of the recommendations concerning the co-administration of antacids, H2-receptor antagonists and proton pump inhibitors. For most patients, proton pump inhibitors should be avoided during sofosbuvir/velpatasvir treatment. If considered necessary, sofosbuvir/velpatasvir should be given with food and taken 4 h before the proton pump inhibitor (at maximum dose comparable to omeprazole 20 mg).
While no dose adjustment of sofosbuvir and ledipasvir is required for patients with mild or moderate renal impairment, the safety of the sofosbuvir-ledipasvir combination has not been assessed in patients with severe renal impairment (eGFR <30 ml/min/1.73 m2) or ESRD requiring haemodialysis. Relative to patients with normal renal function (eGFR >80 ml/min/1.73 m2), the sofosbuvir AUC was 61%, 107% and 171% higher in patients with mild, moderate and severe renal impairment, while the GS-331007 AUC was 55%, 88% and 451% higher, respectively. Thus, no dose adjustment is required for patients with mild or moderate renal impairment, but no dose recommendation can currently be given for patients with severe renal impairment (eGFR <30 ml/min/1.73 m2) or with ESRD.
Since the combination contains ledipasvir and sofosbuvir, any interactions identified with the individual drugs will apply to the combination. The potential (limited) interactions with sofosbuvir have been previously outlined. Since both ledipasvir and sofosbuvir are transported by intestinal P-gp and breast cancer resistance protein (BCRP), any co-administered drugs that are potent P-gp inducers will decrease not only sofosbuvir but also ledipasvir plasma concentrations, leading to reduced therapeutic effect. Although co-administration with drugs that inhibit P-gp and/or BCRP may increase the exposure of sofosbuvir and ledipasvir, clinical consequences are unlikely. One area of focus for ledipasvir interactions is the inhibition of P-gp and/or BCRP whereby ledipasvir may increase the intestinal absorption of co-administered drugs. Thus, caution is warranted with well-studied P-gp substrates such as digoxin and dabigatran, but also potentially with other drugs which are, in part, transported by these proteins (e.g. aliskiren, amlodipine, buprenorphine, carvedilol, cyclosporine). Co-administration of amiodarone with sofosbuvir/ledipasvir is contraindicated due to a serious risk of symptomatic or even fatal bradycardia or asystole (see above, mechanism of interaction is unknown). The use of rosuvastatin is also not recommended (thought to be due to inhibition of hepatic organic anion-transporting protein [OATP] by ledipasvir) and interactions with other statins cannot be excluded. It is important to monitor carefully for statin adverse reactions. Since ledipasvir solubility decreases as pH increases, drugs that increase gastric pH (antacids, H2-receptor antagonists, proton pump inhibitors) are likely to decrease concentrations of ledipasvir. H2-receptor antagonists can be given simultaneously or 12 h apart at a dose not exceeding that equivalent to famotidine 40 mg and proton pump inhibitors simultaneously at a dose comparable to omeprazole 20 mg. Real-world data have suggested slightly reduced SVR rates in patients receiving high-dose proton pump inhibitors, reinforcing the need for caution in patients on such drugs who are treated with sofosbuvir and ledipasvir [31].
Since elbasvir and grazoprevir are substrates of CYP3A and P-gp, inducers of these proteins such as efavirenz, etravirine, phenytoin, carbamazepine, bosentan, modafinil and St John's wort may cause a marked decrease in plasma exposure of both DAAs and are therefore contraindicated. Strong inhibitors of CYP3A (e.g. boosted protease inhibitors, azole antifungals), which may markedly increase plasma concentrations, are either contraindicated or not recommended. In addition to inhibition of CYP3A, grazoprevir plasma concentrations may also be markedly increased by inhibitors of OATP1B1 (including boosted protease inhibitors, cobicistat, cyclosporin, single dose rifampicin). However, there is no effect of acid-reducing agents on the absorption of either DAA.
The potential for grazoprevir/elbasvir to affect other medications is relatively low, although grazoprevir is a weak CYP3A inhibitor (approximately 30% increase in midazolam exposure) and elbasvir a weak inhibitor of P-gp. There needs to be some caution when co-administering drugs that use CYP3A and P-gp in their disposition (e.g. tacrolimus, some statins, dabigatran, ticagrelor).
Based on the findings above, there are limitations on which antiretrovirals can be co-administered with elbasvir/grazoprevir. Currently the antiretrovirals that can be used are the nucleotide reverse transcriptase inhibitors abacavir, lamivudine, tenofovir (either as tenofovir disoproxil fumarate or as tenofovir alafenamide), emtricitabine, rilpivirine, raltegravir, dolutegravir and maraviroc (Table 4A).
Daclatasvir is a substrate of CYP34A and a substrate and inhibitor of P-gp. In addition, it is an inhibitor of OATP1B1 and BCRP. Co-administration of daclatasvir with drugs that strongly induce CYP3A4 and P-gp and thus reduce daclatasvir exposure is contraindicated. This includes anticonvulsants (carbamazepine, phenytoin, oxcarbazepine, phenobarbital), antimycobacterials (rifampicin, rifabutin, rifapentine), systemic dexamethasone and St John's wort. Strong inhibitors of CYP3A4 increase the plasma levels of daclatasvir; therefore, dose adjustments of daclatasvir are recommended. The dose of daclatasvir should be reduced to 30 mg once daily with atazanavir/ritonavir and cobicistat-containing antiretroviral regimens. In contrast, recent data suggest that no dose adjustment is necessary with either darunavir/ritonavir, darunavir/cobicistat or lopinavir/ritonavir. In the ALLY-2 study in HIV coinfected patients receiving sofosbuvir and daclatasvir, patients on a darunavir-based regimen who had daclatasvir dose reduced to 30 mg (based on the original atazanavir/ritonavir study data) had a reduced SVR12, particularly in the 8 week treatment arm, pointing to the need for the standard dose of daclatasvir in patients on this boosted protease inhibitor. With efavirenz (an enzyme inducer), the dose of daclatasvir is recommended to be increased to 90 mg. Due to a lack of data, the same is not recommended with etravirine and nevirapine, both enzyme inducers. There are no drug interactions with tenofovir, emtricitabine, abacavir, lamivudine, zidovudine, stavudine, rilpivirine, raltegravir, dolutegravir or maraviroc.
The dose of daclatasvir should also be reduced to 30 mg with the antibacterials clarithromycin, telithromycin, erythromycin and the antifungals ketoconazole, itraconazole, posaconazole and voriconazole. Studies have been performed with acid-reducing agents (famotidine, omeprazole), escitalopram and an oral contraceptive with no dose adjustment of daclatasvir or the co-medication. However, due to daclatasvir inhibiting some transport proteins, monitoring is required with dabigatran and digoxin and other P-gp substrates.
|
|
| |
| |
|
|
|