| |
Integrase inhibitor versus protease inhibitor based regimen for HIV-1 infected
women (WAVES): a randomised, controlled, double-blind, phase 3 study
|
| |
| |
Download the PDF here
Download the PDF here
"We did the first antiretroviral trial to enroll only women and aimed to assess safety and efficacy of two approved HIV-1 regimens, the single-tablet integrase inhibitor regimen containing elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate compared with the protease inhibitor regimen of ritonavir-boosted atazanavir with emtricitabine and tenofovir disoproxil fumarate.
⋅ The Women AntiretroViral Efficacy and Safety study (WAVES) is an international, randomised, controlled, double-blind, phase 3 study done at 80 sites from Belgium, Dominican Republic, France, Italy, Mexico, Portugal, Puerto Rico, Russia, Thailand, Uganda, the UK, and the USA.
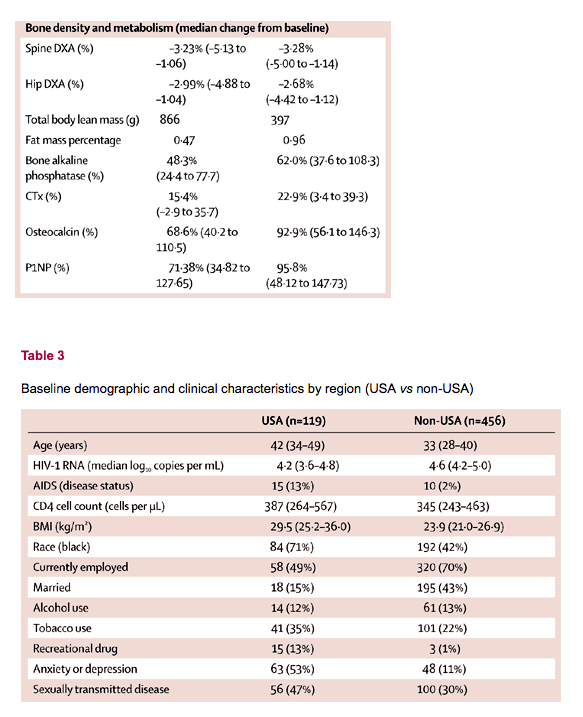
⋅ Most women had attended primary school only. Around two-thirds of women worked outside the home. Most study participants were from the USA, Uganda, and Russia (table 2); of the 103 not from these countries, 56 were from Europe, 23 from the Dominican Republic or Mexico, and 24 from Thailand. Women from the USA differed from the overall study population in several characteristics (table 3), including self-reported histories of anxiety or depression, previous sexually transmitted disease (STD), and recreational drug use (all p<0⋅0001).
⋅ Study drug adherence was also assessed and regional differences in the proportion of participants with at least 95% study drug adherence rate through to week 48 were noted: 61% in the USA, 83% in Uganda, 92% in Russia, and 80% in all other."
⋅ "In addition, the study assessed changes in bone density and body fat, important issues for long-term health. Notably, trunk fat increases, as measured by dual energy x-ray absorptiometry, were greater in women randomised to the protease inhibitor based regimen compared with those receiving the integrase inhibitor regimen. This is by contrast with previous results in which raltegravir—another integrase inhibitor—was shown to have similar amounts of fat gain compared with atazanavir.8 Whether the lower fat gain is a specific advantage of elvitegravir will require a direct comparison and it will be important that these comparisons are done among women as well as men." from Commentary by Judith Currier, Shahin Lockman pdf attached
⋅ "Bone turnover markers specific to bone formation (bone-specific alkaline phosphatase, osteocalcin, and N-terminal propeptide of type 1 procollagen) and markers specific to bone resorption (C-terminal telopeptide of type 1 collagen) were also measured (table 4). Overall, percent change from baseline for all bone markers were lower in the integrase inhibitor group than in the protease inhibitor group (p<0⋅050). An increase in median change in total body lean mass was noted for the integrase inhibitor group (866 g, p=0⋅0004, n=116) versus no significant change in the in the protease inhibitor group (397 g, p=0⋅077, n=126). By contrast, no increase in median total body fat mass for the integrase inhibitor group was noted (945 g, p=0⋅080, n=116) compared with a significant increase in the protease inhibitor group (1412 g, p=0⋅015, n=126). The median increase in body-mass index was 0⋅43 kg/m2 in both groups (p=0⋅50)."
ICAAC/2015: Elvitegravir (EVG)/Cobicistat (COBI)/Emtricitabine (FTC)/Tenofovir Disoproxil Fumarate (TDF) Is Superior to Ritonavir (RTV)-Boosted Atazanavir (ATV) Plus FTC/TDF in Treatment-Naïve Women With HIV-1 Infection (WAVES Study) - (09/21/15)
--------------------------
Integrase inhibitor versus protease inhibitor based regimen for HIV-1 infected women (WAVES): a randomised, controlled, double-blind, phase 3 study
Lancet HIV May 27 2016
Kathleen Squires, Cissy Kityo, Sally Hodder, Margaret Johnson, Evgeny Voronin, Debbie Hagins, Anchalee Avihingsanon, Ellen Koenig,
Shuping Jiang, Kirsten White, Andrew Cheng, Javier Szwarcberg, Huyen Cao
Summary
Background
Women are under-represented in HIV antiretroviral therapy (ART) studies. Guidelines for selection of ART as initial therapy in patients with HIV-1 infection do not contain sex-specific treatment. We aimed to assess the safety and efficacy of the single tablet integrase inhibitor regimen containing elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate compared with a boosted protease inhibitor regimen of ritonavir-boosted atazanavir with emtricitabine and tenofovir disoproxil fumarate.
Methods
In this international, randomised, controlled, double-blind, phase 3 study (Women AntiretroViral Efficacy and Safety study [WAVES]), we recruited treatment-naive HIV-infected women with an estimated creatinine clearance of 70 mL/min or higher from 80 centres in 11 countries. Women were randomly assigned (1:1) to receive elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate (integrase inhibitor regimen) or ritonavir-boosted atazanavir with emtricitabine and tenofovir disoproxil fumarate (protease inhibitor based regimen); regimens were masked with matching placebos.
Randomisation was done by a computer-generated allocation sequence (block size four) and was stratified by HIV-1 RNA viral load and race. Investigators, patients, study staff, and those assessing outcomes were masked to treatment group. All participants who received one dose of study drug were included in the primary efficacy and safety analyses. The main outcome was the proportion of patients with plasma HIV-1 RNA less than 50 copies per mL at week 48 as defined by US Food and Drug Administration snapshot algorithm (prespecified non-inferiority margin of 12%). This study is registered with ClinicalTrials.gov, number NCT01705574.
Findings
Between Nov 28, 2012, and March 12, 2014, 575 women were enrolled. 289 were randomly assigned to receive the integrase inhibitor regimen and 286 to receive the protease inhibitor based regimen. 252 (87%) women in the integrase inhibitor group had plasma HIV-1 RNA less than 50 copies per mL at week 48 compared with 231 (81%) women in the protease inhibitor group (adjusted difference 6⋅5%; 95% CI 0⋅4-12⋅6). No participant had virological failure with resistance in the integrase inhibitor group compared with three participants ([1%]; all Met184Val/Ile) in the protease inhibitor group. 19 women in the protease inhibitor group discontinued because of adverse events compared with five in the integrase inhibitor group.
Interpretation
WAVES shows that clinical trials of ART regimens in global and diverse populations of treatment-naive women are possible. The findings support guidelines recommending integrase inhibitor based regimens in first-line antiretroviral therapy.
Introduction
Half of the cases of HIV worldwide are in women, and the number of women acquiring HIV infection continues to rise.1 Research guidelines have long advocated for sex-based assessment of drug efficacy, toxicity, and tolerability profiles;2, 3 but women continue to be under-represented in clinical trials assessing efficacy and safety of antiretroviral treatment (ART) among HIV-1 infected people. One of the consequences of this restricted representation is the absence of definitive information about the specific efficacy and safety of ART in women.4, 5, 6, 7, 8, 9 The selection of ART should be evidence based and take into account several factors, including regimen potency, side-effects, level of adherence required for efficacy, and quality of life specific to the population of patients. Current guidelines for first-line treatment of HIV-1 infection include the use of two nucleoside reverse transcriptase inhibitors (NRTIs) plus a third active drug from a different class.10, 11, 12 The integrase strand transfer inhibitor (elvitegravir, 150 mg) coformulated with cobicistat (150 mg), emtricitabine (200 mg), and tenofovir disoproxil fumarate (300 mg) in a single-tablet regimen is a preferred ART regimen in treatment-naive patients and atazanavir (300 mg) boosted by ritonavir (100 mg) plus a preferred two-NRTI backbone (emtricitabine plus tenofovir disoproxil fumarate) is well tolerated in HIV-infected women13 and remains a preferred regimen during pregnancy.10, 11, 14
We did the first antiretroviral trial to enrol only women and aimed to assess safety and efficacy of two approved HIV-1 regimens, the single-tablet integrase inhibitor regimen containing elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate compared with the protease inhibitor regimen of ritonavir-boosted atazanavir with emtricitabine and tenofovir disoproxil fumarate.
Research in context
Evidence before this study
We searched PubMed for reports of large randomised clinical trials assessing antiretroviral treatment in ART-naive women and found no studies. Search terms included "HIV, "naive" AND "women" or "female" AND "antiretroviral" AND "randomized trial" and searches were limited to articles published in English between Jan 1, 1997, and Dec 31, 2015. Women account for half of the global HIV epidemic yet remain under-represented in HIV clinical trials. Current HIV treatment guidelines are based on data obtained mainly from men and might promote sex bias and inaccuracies in the paradigm of evidence-based medicine. To our knowledge, there are no published data from randomised clinical studies that focus primarily on antiretroviral treatment in women.
Added value of this study
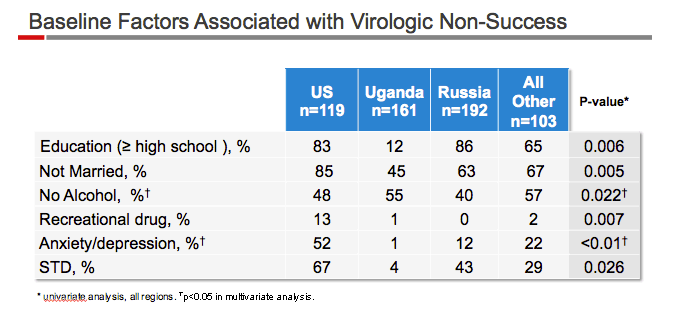
This first all-women, randomised, double-blind clinical trial compared two approved ART regimens: integrase based (elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate) and protease inhibitor based (ritonavir-boosted atazanavir with emtricitabine and tenofovir disoproxil fumarate). The integrase inhibitor group had superior efficacy to the protease inhibitor based, differing from previous clinical trials (with mostly male participants) in which the integrase inhibitor group was non-inferior in efficacy to the protease inhibitor group. Unanticipated regional differences in efficacy and tolerability were noted. The highest virological response was reported in Ugandan women and the lowest rate of viral suppression was seen in the USA, regardless of treatment group.
Interpretation
The WAVES study is the only completed randomised clinical trial to date done exclusively among women. The WAVES population was geographically and ethnically diverse, providing a better understanding of several factors that might affect clinical outcome. The randomised study groups were well matched and the outcome data indicate that in the setting of this clinical trial the integrase inhibitor group provided superior efficacy with increased tolerability, offering new insights and treatment information to clinicians caring for women with HIV infection.
Methods
Study design and participants
The Women AntiretroViral Efficacy and Safety study (WAVES) is an international, randomised, controlled, double-blind, phase 3 study done at 80 sites from Belgium, Dominican Republic, France, Italy, Mexico, Portugal, Puerto Rico, Russia, Thailand, Uganda, the UK, and the USA. Women aged 18 years or older were eligible if they were HIV-1 infected had not received previous ART, had plasma HIV-1 RNA viral load 500 copies per mL or greater, and an estimated glomerular filtration rate of at least 70 mL/min. Additional inclusion criteria included aspartate and alanine aminotransferase concentration below five times the upper limit of normal, total bilirubin 1⋅5 mg/dL or less, or a normal direct bilirubin, and sensitivity to emtricitabine, tenofovir disoproxil fumarate, and atazanavir at screening. Exclusion criteria included current pregnancy or breastfeeding. Women who became pregnant during the study had the option to continue unblinded study ART after providing additional informed consent.
This study was approved by the institutional review board or independent ethics committee at each participating site and was done in compliance with the principles of the Declaration of Helsinki, Good Clinical Practice guidelines, and local regulatory requirements. The study was designed and done according to the protocol by the funder (Gilead Sciences) in collaboration with the investigators. All patients provided written informed consent. An independent data and safety monitoring committee met regularly to review the progress of the study
Randomisation and masking
Eligible women were randomly assigned (1:1) to receive elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate (integrase inhibitor regimen) or ritonavir-boosted atazanavir with emtricitabine and tenofovir disoproxil fumarate (protease inhibitor based regimen), as well as a matching placebo based on treatment randomisation. Investigators, patients, and study staff providing treatment, assessing outcome, and collecting data were masked to the assigned treatment group. A computer-generated allocation sequence was created by Bracket (San Francisco, CA, USA), and block randomisation (block size of four) was stratified by HIV-1 RNA concentration (≤100 000 copies per mL, >100 000 to ≤400 000 copies per mL, or >400 000 copies per mL) and race (black or non-black). Study investigators determined eligibility, obtained a participant number, and received automated treatment assignment based on a randomisation sequence.
Procedures
In addition to laboratory and clinical tests to assess eligibility, the screening assessments included medical and gynaecological history and social demographic information including specific questions on drug and alcohol use, employment, education, marital status, and number of children living in the household. After the screening and baseline visits, enrolled women returned to the clinic every 4 weeks until week 16 and then every 8 weeks until week 48. At all study visits, assessments were done for adverse events and concomitant medications, and complete or symptom-directed physical examinations were done. Laboratory tests included haematological analysis, serum chemistry, fasting lipid parameters, CD4 cell counts, measures of bone turnover (C-terminal cross-laps, osteocalcin, N-terminal propeptide, and bone alkaline phosphatase [Covance Laboratories, Indianapolis, IN, USA]), and measures of HIV-1 RNA (Roche TaqMan version 2.0, Roche Diagnostics, Rothkreuz, Switzerland). Questionnaires including the HIV Treatment Satisfaction Questionnaire, Short Form Health Survey, and self-reported adherence form were administered throughout the study. As part of the screening requirements, study samples were analysed for pre-existing resistance in the protease and reverse transcriptase portions of the pol gene with the GeneSeq assay (Monogram Biosciences, South San Francisco, CA, USA). Resistance analyses of protease, reverse transcriptase, and integrase were done on plasma samples from women who were on study drugs and had either suboptimal virological response (confirmed HIV-1 RNA ≥50 copies per mL and <1 log10 reduction from baseline at the week 8 visit), or virological rebound (two consecutive visits with HIV-1 RNA ≥400 copies per mL after achieving HIV-1 RNA <50 copies per mL, or as having two consecutive visits with >1 log10 increase in HIV-1 RNA from nadir).
Additionally, resistance analysis was done in women who were on study drugs, had not been analysed previously, and who had HIV-1 RNA ≥400 copies per mL at week 48 or their last visit (at or after week 8). Subsequent to the first resistance testing, participants who had repeated confirmed virological failure were assessed for resistance retesting on a case-by-case basis, at the funder's or investigator's discretion.
Dual energy x-ray absorptiometry scans were done in a subset of women who agreed to participate in the DXA substudy, before study drug administration at baseline and at week 48 (BioClinica, Newton, PA, USA). Percent bone mineral density changes from baseline at the lumbar spine and hip were calculated. Total body fat and lean mass changes were measured with whole body dual energy x-ray absorptiometry at baseline and week 48. Samples of plasma, hair, and cervicovaginal fluid were obtained in a subset of women for pharmacokinetic studies.
Outcomes
The prespecified primary efficacy endpoint was the proportion of participants with a plasma HIV-1 RNA viral loads of less than 50 copies per mL at week 48, as determined with the use of the US FDA-defined snapshot algorithm,15 now widely used in the analysis of HIV trials. The secondary endpoints included the change from baseline in the CD4 cell count and the safety profile through to week 48. Safety assessments included standard laboratory testing and adverse event, coded with the MedDRA.
Statistical analysis
All women who were randomly assigned a treatment and who had received at least one dose of the study drug were included in the primary endpoint analysis (intent-to-treat analysis set). Baseline characteristics were summarised with descriptive statistics. For categorical data, p values were calculated from the Cochran-Mantel-Haenszel test (general association statistic was used for nominal data, row mean scores differ statistic was used for ordinal data). For continuous data, p value was from the two-sided Wilcoxon rank sum test. For the primary endpoint, the percentage differences and the associated 95% CIs were computed with the baseline HIV-1 RNA concentration and race stratum adjusted Mantel-Haenszel proportions. In the snapshot analysis of full intention-to-treat, women who were still on study treatment with HIV-1 RNA less than 50 copies per mL in the week 48 window (between days 309 and 378) were classified as a virological success. The following were classified as virological non-success: women with HIV-1 RNA of 50 or more copies per mL, women with missing HIV-1 RNA data in the week 48 window, or women who changed treatment before week 48.
The non-inferiority of the integrase inhibitor regimen compared with the protease inhibitor based regimen would be concluded if the lower bound of the two-sided 95% CI is greater than −12%. The 95% CI was calculated based on stratum-adjusted Mantel-Haenszel proportions. A sample size of 255 per treatment group provided a 95% power to detect a non-inferiority margin of 12% at week 48. On the basis of previous clinical trial data in women, the assumed response rate was 83% per group. The one-sided significance level was set at 0⋅025.
Upon establishment of non-inferiority, the superiority of the integrase inhibitor regimen over the protease inhibitor based regimen was also assessed. The same 95% CI assessing non-inferiority was used to assess superiority with a prespecified margin of 0, where the lower bound of the 95% CI greater than 0 would conclude that the integrase inhibitor regimen is superior to the protease inhibitor based regimen. Two-sided Cochran-Mantel-Haenszel test was also used to assess superiority in data stratified by baseline HIV-1 RNA viral load and race. A prespecified subgroup analysis of treatment differences was done on the basis of baseline HIV-1 RNA viral loads and CD4 count, age, race, and study drug adherence. Adherence to the study drug was calculated as number of pills taken (as measured by pill counts conducted at each study visit) divided by number of pills prescribed.
Changes from baseline in CD4 cell count at week 48 were summarised with descriptive statistics. Difference in changes from baseline in CD4 cell count between treatment groups and the associated 95% CIs were calculated by analysis of variance models, including baseline HIV-1 RNA counts, race, and treatment group as fixed effects in the model.
The safety analysis set included all randomly assigned patients who received at least one dose of study drugs. Safety data in the safety analysis set were analysed with descriptive statistics. Adverse events were coded with the Medical Dictionary for Regulatory Activities (MedDRA).
The study was done according to protocol without significant deviations and is registered with ClinicalTrials.gov, number NCT01705574.
Role of the funding source
Gilead Sciences funded and monitored the study, collected and analysed the data, interpreted the results, and helped to write the report. KS, CK, and SH had full access to the data, interpreted the results, and helped to write the report. All authors had full access to the data, could request additional analyses, and could provide input into the interpretation of results. AC, JS, and HC made the decision to submit the report for publication.
Results
Between Oct 24, 2012, and Jan 28, 2014, 810 women were screened for eligibility. Of the 227 who did not meet the study entry eligibility criteria (screened participants could have more than one inclusion or exclusion criterion), 129 (57%) women were from Uganda. The most common screen fail reasons for the Uganda site include: eGFR less than 70 mL/min (64%), HIV-1 RNA less than 500 copies per mL (14%), abnormal haematology profile (11%), positive serum pregnancy (4%), screening genotype with resistance study drugs (4%), withdrew consent (4%), and loss to follow-up (3%). Of the remaining screen failures at other sites, reasons included: HIV-1 RNA less than 500 copies per mL (19%), screening genotype with resistance study drugs (6%), eGFR less than 70 mL/min (27%), abnormal haematology profile (13%), positive serum pregnancy (1%), substance abuse (4%), did not agree to protocol recommended contraceptive methods (3%), previous ART (4%), other ongoing serious clinical conditions or history of recent malignant disease (2%), contraindicated medication (2%), and other clinical conditions that in opinion of investigator, would make participants unsuitable for study (5%).
Between Nov 28, 2012, and March 12, 2014, 575 women met eligibility crtieria and were enrolled; of whom, 289 were randomly assigned to the integrase inhibitor regimen and 286 to the protease inhibitor based regimen (figure 1). The demographic and baseline clinical characteristics were generally balanced between the two groups (table 1). Median age of study participants was 35 years, 276 (48%) of 575 participants were black, and 449 (78%) had asymptomatic HIV disease. The median HIV-1 RNA viral load at baseline was 4⋅51 log10 copies per mL, median CD4 count was 358 cells per μL; 16 (3%) of 477 (due to availability of samples) were positive for hepatitis B surface antigen and 47 (9%) of 531 (due to availability of samples) were positive for hepatitis C antibody.
Unprotected heterosexual intercourse was the leading method of HIV-1 acquisition among the participants. At the time of enrolment, 344 (60%) of 575 women reported being sexually active, 308 (90%) of whom were in a monogamous relationship. The use of contraceptive methods was well matched between groups (table 1). 481 (84%) of 575 women reported previous pregnancy and 53% had children younger than 18 years living in the household (table 1). Most women had attended primary school only. Around two-thirds of women worked outside the home. Most study participants were from the USA, Uganda, and Russia (table 2); of the 103 not from these countries, 56 were from Europe, 23 from the Dominican Republic or Mexico, and 24 from Thailand. Women from the USA differed from the overall study population in several characteristics (table 3), including self-reported histories of anxiety or depression, previous sexually transmitted disease (STD), and recreational drug use (all p<0⋅0001).
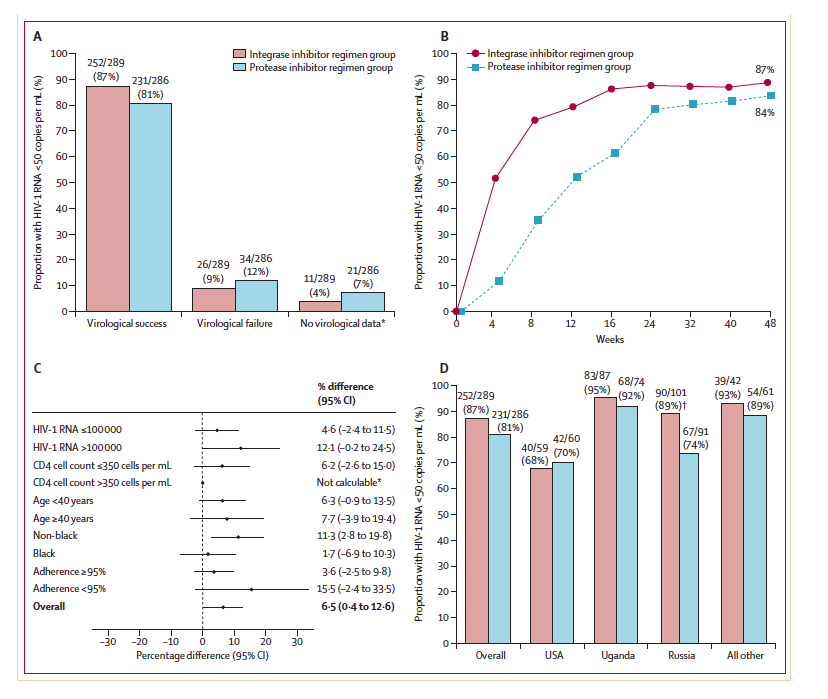
By week 48, 252 (87%) of 289 patients in the integrase inhibitor group and 231 (81%) of 286 in the protease inhibitor group had virological response: adjusted treatment difference 6⋅5% (95% CI 0⋅4-2⋅6; figure 2). The finding shows non-inferiority and the lower bound of the 95% CI being above 0, establishes superior efficacy for the integrase inhibitor regimen. The virological response rate was significantly higher in the integrase inhibitor group than in the protease inhibitor group (p=0⋅034, figure 2). The response rates were consistent across the analysed subgroups and generally favoured the integrase inhibitor regimen (figure 2). The most notable treatment effect was noted in the non-black subgroup, with response rates of 89% in women receiving the integrase inhibitor regimen and 78% in those receiving the comparator (adjusted treatment difference 11⋅3%; 95% CI 2⋅8-19⋅8).
The efficacy outcome also differed by regions (figure 2). Efficacy of the protease inhibitor regimen was significantly lower than that of the integrase inhibitor in Russia. Study drug adherence was also assessed and regional differences in the proportion of participants with at least 95% study drug adherence rate through to week 48 were noted: 61% in the USA, 83% in Uganda, 92% in Russia, and 80% in all other.
26 participants (9%) in the integrase inhibitor group and 34 (12%) in the protease inhibitor group had virological failure. 12 women in each group were lost to follow-up (11 in the USA, five in Russia, four in Uganda, two in Portugal, one in Italy, and one in Thailand). Five women in the integrase inhibitor group discontinued due to adverse events compared with 19 in the protease inhibitor group (table 4). CD4 counts increased in both groups; at week 48 the mean count increase was 221 cells per μL (SD 165⋅1) in the integrase inhibitor group and 212 cells per μL (SD 176⋅8) in the protease inhibitor group.
Figure 2: Efficacy data up to week 48
(A) Proportion of participants who were responders (HIV-1 RNA viral load of less than 50 copies per mL at week 48, according to the US Food and Drug
Administration snapshot algorithm), had virological failure, or had no virological data. (B) Proportion of participants with HIV-1 RNA load of less than
50 copies per mL according to study visit through to week 48. (C) Diff erence in response rates in the subgroups from the intention-to-treat population; all
comparisons are represented as adjusted diff erences in proportion (integrase inhibitor regimen minus the protease inhibitor based regimen); 95% CIs calculated after adjustment by baseline HIV-1 RNA and race strata. (D) shows the proportion of participants who are responders by regions by treatment group. *Data not calculable in subgroup CD4 count 350 cells per μL or greater due to imbalance in race and viral load distribution in the subgroup. †Statistical signifi cance (p=0.0072).
Baseline genotypic analysis showed evidence of transmitted resistance substitutions (30% across both groups). 20% had resistance to non-nucleoside reverse transcriptase inhibitors, 15% to nucleoside reverse transcriptase inhibitors, and 1⋅9% to protease inhibitors. Baseline phenotypic analysis showed a lower level of pre-existing resistance substitutions on predicted phenotypic resistance to antiretroviral drugs: rilpivirine (7⋅0%), nevirapine and etravirine (5⋅2% each), efavirenz (4⋅9%), and zidovudine (1⋅0%). As required by the protocol, all participants at baseline had HIV-1 that was fully sensitive to study drugs. Subtyping analysis showed higher prevalence of non-B subtype HIV-1 (74%) than subtype B (26%). The most frequent non-B subtypes were A or A1 (46%), D (8%), C (5%), AE (5%), or AG (5%; table 2). The resistance analysis was done in seven women (2%) in the integrase inhibitor group and 12 women (4%) in the protease inhibitor group. No women developed virological failure with genotypic or phenotypic resistance to study drugs in the integrase inhibitor group compared with three (1%]; all Met184Val/Ile) in the protease inhibitor group.
Most adverse events were reported as mild (grade 1) or moderate (grade 2) in severity (table 4). Serious adverse events were reported in 24 women (8%) in the integrase inhibitor group and 29 women (10%) in the protease inhibitor group and no deaths occurred in either group. Five women (2%) in the integrase inhibitor group and 19 (7%) in the protease inhibitor group discontinued due to adverse events (table 4). Reasons for discontinuation in the integrase inhibitor group included rash with nausea and rash with jaundice, alanine transferase elevation, dyspepsia, peptic ulcer disease, and tuberculosis. Of the 19 adverse event-associated discontinuations in the protease inhibitor group, four were attributed to either jaundice or an increased in serum bilirubin, nine to skin-related disorders (rash with or without additional adverse events), and two to renal events (acute renal failure and abnormal eGFR). Jaundice and icterus were both more common in the protease inhibitor group than in the integrase inhibitor group (table 4).
Treatment-emergent laboratory abnormalities were mild or moderate in severity in the integrase inhibitor group and of greater severity and frequency in the protease inhibitor group (table 4). 64 women (23%) in the integrase inhibitor group and 171 (61%) in the protease inhibitor group had severe and life-threatening laboratory abnormalities (grade 3 or 4); the difference was largely in bilirubin abnormalities: hyperbilirubinaemia affected two (1%) in integrase inhibitor group and 130 (46%) in the protease inhibitor group; table 4). Increases from baseline for metabolic measures did not differ substantially between groups, except for total cholesterol; however, the total cholesterol to high-density lipoprotein (HDL) ratio was similar between the groups (table 4); and the median change in total cholesterol to HDL ratio was −0⋅1 in both groups. Small changes in serum creatinine concentration were seen in both groups and the median change (IQR) from baseline in serum creatinine at week 48 was higher in the integrase inhibitor group (p=0⋅030) and changes in eGFR were not significantly different (p=0⋅15). Median percent decreases in bone mineral density were similar for the integrase inhibitor group versus the protease inhibitor group (table 4). Bone turnover markers specific to bone formation (bone-specific alkaline phosphatase, osteocalcin, and N-terminal propeptide of type 1 procollagen) and markers specific to bone resorption (C-terminal telopeptide of type 1 collagen) were also measured (table 4). Overall, percent change from baseline for all bone markers were lower in the integrase inhibitor group than in the protease inhibitor group (p<0⋅050). An increase in median change in total body lean mass was noted for the integrase inhibitor group (866 g, p=0⋅0004, n=116) versus no significant change in the in the protease inhibitor group (397 g, p=0⋅077, n=126). By contrast, no increase in median total body fat mass for the integrase inhibitor group was noted (945 g, p=0⋅080, n=116) compared with a significant increase in the protease inhibitor group (1412 g, p=0⋅015, n=126). The median increase in body-mass index was 0⋅43 kg/m2 in both groups (p=0⋅50).
Women enrolled in this study were required to use two forms of birth control, and pregnancy testing occurred at every study visit. Women who became pregnant during the study had the option to continue unblinded study ART. 24 women became pregnant (26 pregnancies), and 16 elected to continue study drugs (eight in the integrase inhibitor group and eight in the protease inhibitor group [two pregnancies reported in one participant]). Spontaneous abortion in the first trimester occurred in four of the 16 women (two in the integrase inhibitor group and two in the protease inhibitor group). Uncomplicated term delivery was confirmed for 12 pregnancies with no congenital malformations reported. Virological suppression was confirmed in 12 of these women at week 48. One woman (in the integrase inhibitor group) had rebound viraemia at week 48 (HIV-RNA 14 500 copies per mL) but subsequently had virological suppression at the time of delivery.
Discussion
The integrase inhibitor regimen (elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate) had superior efficacy to the protease inhibitor based regimen (atazanavir, ritonavir, emtricitabine, and tenofovir disoproxil fumarate). High virological responses were noted in most regions and across different HIV subtypes. No emergent resistance was detected in the integrase inhibitor group but was present in three women in the protease inhibitor group.
The higher response rate in this all-women study differs from previous clinical trials (with mostly male participants) in which the protease inhibitor based regimen was non-inferior, but not superior, in efficacy to the integrase inhibitor group in two large randomised trials.4, 16, 17, 18 Superiority of the integrase inhibitor group in this study was mainly driven by the higher rate of discontinuation in the protease inhibitor group, than in the integrase inhibitor group, because of adverse events (mainly rash and bilirubin-associated adverse events). The discontinuation rates in this study were higher than that previously observed for men.13, 19
A higher virological failure rate among women receiving ritonavir-boosted atazanavir compared with efavirenz has been previously shown in ACTG 5202, a randomised trial comparing abacavir plus lamivudine with emtricitabine plus tenofovir disoproxil fumarate and either efavirenz or atazanavir (boosted with ritonavir).25 Women randomly assigned ritonavir-boosted atazanavir had more than double the virological failure rate compared with those assigned efavirenz; virological failures among men were similar in both groups.25 Pharmacokinetic studies in ACTG 5202 showed higher atazanavir exposure among women than in men; however, diferences in safety or tolerability between the sexes were not significant. Virological response rates were lower in women in the randomised trial comparing ritonavir-boosted atazanavir with ritonavir-boosted lopinavir,20 in which 67% of women in the atazanavir plus group had HIV-1 RNA <50 copies per mL compared with 77% of men at week 96. On-treatment analysis failed to confirm sex differences in response rates and a similar pattern was seen in the lopinavir group. The protease inhibitor based regimen was chosen as a comparator regimen in this sudy because it was a preferred US Department of Health & Human Services regimen at the time of study inititiation and remains a preferred regimen for pregnant women.
In this study, lower response rates in the ritonavir-boosted atazanavir group were driven by study-drug discontinuations for rash and bilirubin-related adverse events. The rate of discontinuation caused by rash was high compared with historical rates reported for men, showing sex differences in reported severities of adverse events associated with antiretroviral drugs.13, 19, 21, 22, 23 Pharmacokinetic studies are pending and will delineate whether increased atazanavir concentrations were associated with study-drug discontinuation.
Although tenofovir disoproxil fumarate is generally well tolerated, patients with kidney disease or those who are receiving concomitant ritonavir-boosted protease inhibitors are at increased risks for renal events.24, 25 Renal adverse events leading to discontinuation were rare in this study. The median increases from baseline in serum creatinine and eGFR were lower than those reported in previous clinical studies;26, 27 reasons for the lower observed rate are unknown.
Moderate decreases in bone mineral density occurred after initiation of antiretroviral regimens and eventually stabilised;28 and similar changes in bone mineral density were noted between the two treatment groups. Changes in bone resorption and formation were smaller in the integrase inhibitor group. These findings have important clinical implications because ritonavir-boosted protease inhibitors are often favoured in women of childbearing potential and an atazanavir based regimen is a standard regimen for pregnant women.
Unanticipated regional differences in efficacy were noted. The highest virological response was observed in Ugandan women and the lowest rate of viral suppression was seen in the USA, regardless of treatment group. The relatively low viral suppression rate among US women was associated with the lowest rate of study drug adherence and a high rate of loss to follow-up. High rates of virological failure among women in previous clinical trials have been attributed to complex socioeconomic factors leading to poor medication adherence.4, 29 Although the exact predictor of health and virological outcome is complex, women from the USA had a distinct sociodemographic profile compared with non-US women in this study.5, 6, 8, 30 This finding emphasises the reported disproportionally poorer health outcome in HIV-infected women in the USA31, 32, 33 and warrants further research to better understand barriers to effective HIV treatment faced by US women. A second unanticipated regional difference was the lower response rate to the protease inhibitor based regimen in Russia, where disproportionate discontinuation of study drugs was noted. Whether or not the pattern resulted from a lower threshold for discontinuation because of rash or other factors leading to increased incidence of skin events, is unclear. These data highlight the difficulty in assessing the precise factors that affect the efficacy and ART options for women regionally and globally.
This study has several noteworthy limitations. Validated instruments for the population studied were not used in the WAVES study to assess past or present anxiety or depression, previous STDs, and recreational drug use. Therefore, the higher reported prevalence of these disorders among US women compared with other regions might reflect a differential understanding of the survey questions or in willingness to self-report these disorders as well as past recreational drug use. Additionally, the opportunity for diagnosis of anxiety or depression and STDs might differ between regions. Dissimilar discontinuation rates might result from higher predisposition to certain adverse events in some parts of the world; however, varying clinical perceptions and decisions to discontinue study drug could also have contributed to the observed differences. Finally, the two study regimens were assessed only up to 48 weeks.
In conclusion, WAVES is the first randomised clinical trial to assess the efficacy and tolerability of ART in treatment-naive women only, showing that it is feasible to enrol and retain women in clinical trials of antiretroviral efficacy and safety. High virological and immunological responses were achieved in this clinical study. The strength of the study includes the randomised blinded study design in a cohort of women with clinical characteristics reflecting the current epidemiology of HIV infection. Additionally, the WAVES population was geographically and ethnically diverse, providing a better understanding of multiple factors that could affect clinical outcome. The randomised study groups were well matched and the outcome data indicate that in the setting of this clinical trial, the integrase inhibitor group provided superior efficacy with increased tolerability, offering new insights and treatment information to clinicians caring for women with HIV infection.
|
|
| |
| |
|
|
|