| |
Hepatitis B cure: From discovery to regulatory approval - Review
|
| |
| |
Download the PDF here
Jnl of Hepatology Aug 1 2017 - Anna S. Lok1,, Fabien Zoulim2, Geoffrey Dusheiko3, Marc G. Ghany4 1Division of Gastroenterology and Hepatology, University of Michigan, Ann Arbor, MI, United States; 2Cancer Research Center of Lyon- INSERM U1052, Hospices Civils de Lyon, Lyon University, Lyon, France; 3University College London Medical School and Kings College Hospital, London, UK; 4Liver Diseases Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, United States
Summary
The majority of persons currently treated for chronic hepatitis B require long-term or lifelong therapy. New inhibitors of hepatitis B virus entry, replication, assembly, or secretion and immune modulatory therapies are in development. The introduction of these novel compounds for chronic hepatitis B necessitates a standardised appraisal of the efficacy and safety of these treatments and definitions of new or additional endpoints to inform clinical trials. To move the field forward and to expedite the pathway from discovery to regulatory approval, a workshop with key stakeholders was held in September 2016 to develop a consensus on treatment endpoints to guide the design of clinical trials aimed at hepatitis B cure. The consensus reached was that a complete sterilising cure, i.e., viral eradication from the host, is unlikely to be feasible. Instead, a functional cure characterised by sustained loss of hepatitis B surface antigen with or without hepatitis B surface antibody seroconversion, which is associated with improved clinical outcomes, in a higher proportion of patients than is currently achieved with existing treatments is a feasible goal. Development of standardised assays for novel biomarkers toward better defining hepatitis B virus cure should occur in parallel with development of novel antiviral and immune modulatory therapies such that approval of new treatments can be linked to the approval of new diagnostic assays used to measure efficacy or to predict response. Combination of antiviral and immune modulatory therapies will likely be needed to achieve functional hepatitis B virus cure. Limited proof-of-concept monotherapy studies to evaluate safety and antiviral activity should be conducted prior to proceeding to combination therapies. The safety of any new curative therapies will be paramount given the excellent safety of currently approved nucleos(t)ide analogues.
Summary and recommendations
A summary of the recommendations on endpoints and design of clinical trials for HBV cure is presented in Table 2. Improved understanding of the HBV life cycle and host immune response to HBV has facilitated discovery and design of antiviral therapies directed against multiple steps of the HBV replication cycle, as well as potential immunomodulatory therapies to restore immune response to HBV infection. Development of novel HBV therapies will be further aided by the availability of improved cellular and animal infection models. While this progress has raised the possibility of a cure for hepatitis B, a complete sterilising cure, i.e., viral eradication from the host, may be unrealistic due to the presence of integrated HBV DNA. Although HBsAg clearance is infrequent, it does occur with NA and IFN therapy and is associated with improved clinical outcomes. Therefore, a functional cure (characterised by sustained loss of HBsAg with or without anti-HBs seroconversion) after a finite course of novel antiviral and immune modulatory therapies in a higher proportion of patients than is currently achieved with existing treatments is an attainable goal.
Cooperation between academia, industry, and regulatory agencies to standardise and validate surrogate markers for cure, in order to facilitate the development of curative therapies and to facilitate the path from discovery to regulatory approval, is required. Limited proof-of-concept monotherapy studies to evaluate safety and antiviral activity should be conducted prior to proceeding to combination therapy. The safety of any new curative therapies will be paramount given the excellent safety of currently approved NAs. Continued collaboration among the stakeholders will make HBV "cure" a reality for patients with chronic HBV.
Introduction
The advent of several novel antiviral and immune modulatory therapies for chronic hepatitis B now necessitates a standardised appraisal of the efficacy and safety of these therapies, and definitions of new or additional endpoints to inform clinical trials. To move the field forward, and to expedite the pathway from discovery to regulatory approval, the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver jointly organised the Hepatitis B Treatment Endpoints Workshop on September 8-9, 2016, in Alexandria, Virginia. The primary goal of this workshop was to assemble key stakeholders from regulatory agencies (US Food and Drug Administration and European Medicines Agency), biopharmaceutical and biotechnology companies engaged in development of diagnostic tests and therapeutic agents for hepatitis B, and academia in order to develop a consensus on treatment endpoints to guide the design of clinical trials aimed at hepatitis B cure.
Sixty-six (33%) of 202 participants completed a premeeting survey, including four from regulatory agencies, 31 from industry, 28 from academia, and three from other health care sectors. During the workshop, experts reviewed the natural history of chronic hepatitis B virus (HBV) infection, efficacy of currently approved treatments, potential antiviral targets and approaches to restore immune responsiveness to HBV, and preclinical and early-phase clinical trial data on novel antiviral and immune modulatory therapies for chronic HBV. The workshop concluded with a session on the definition of HBV cure; efficacy endpoints, safety assessments, target populations, and design of clinical trials; and diagnostic assays needed to support development of curative therapies. These topics were further discussed during a closed session involving 23 experts (including the four authors) representing all constituent groups. This report summarises the discussions and consensus opinions of the 2-day meeting.
Definition of HBV cure
The goal of developing new therapies is to achieve HBV cure, i.e., elimination of HBV, thereby allowing treatment to be stopped with no risk of virological relapse and no risk of liver disease progression. However, a true cure may not be feasible because HBV DNA is integrated into the host genome; even among persons who have recovered from acute HBV, viral covalently closed circular DNA (cccDNA) can be detected in the liver, explaining the reactivation of HBV replication when these "recovered" persons are profoundly immunosuppressed. However, the observation that hepatitis B surface antigen (HBsAg) may become undetectable in serum after clinical recovery from acute hepatitis B, spontaneously during the course of chronic HBV infection, and during or after nucleosid(t)e analogue (NA) or interferon (IFN) therapy, despite the likelihood of persistent integrated HBV genomes, argues for the feasibility of achieving undetectable levels of HBsAg.
A key objective of the meeting was to establish a definition of cure. Three definitions of HBV cure were proposed at this meeting: (i) complete sterilising cure with undetectable HBsAg in serum and eradication of HBV DNA including intrahepatic cccDNA and integrated HBV DNA; (ii) functional cure with sustained, undetectable HBsAg and HBV DNA in serum with or without seroconversion to hepatitis B surface antibody (anti-HBs) after completion of a finite course of treatment, resolution of residual liver injury, and a decrease in risk of hepatocellular carcinoma (HCC) over time (several levels of functional cure including complete shutdown of cccDNA transcription, elimination of cccDNA, complete resolution of liver damage, and elimination of risk of HCC were discussed); (3) partial cure with detectable HBsAg but persistently undetectable HBV DNA in serum after completion of a finite course of treatment.
The vast majority (87.9%) of survey respondents selected functional cure (sustained HBsAg loss) as the goal for new HBV therapies. This selection was endorsed by other participants and the expert panel as a feasible goal. In addition, functional cure offers several other advantages: it is easy to assess and tests are widely available, it is associated with improved clinical outcomes and lower rates of disease reactivation, and once achieved, there is no further requirement for therapy.
There was less consensus regarding the necessity of achieving anti-HBs seroconversion because it is unclear whether durability of HBsAg loss and clinical benefits of HBsAg loss are dependent on the development of anti-HBs. Importantly, few of the participants believed elimination of cccDNA was a mandatory criterion for functional cure, and less than half required that cccDNA be rendered transcriptionally inactive, reflecting uncertainties over whether new therapies in development can silence or clear cccDNA, as well as pragmatic difficulties in measuring cccDNA.
Some members of the expert panel considered partial cure (sustained suppression of HBV replication off treatment but persistent presence of HBsAg) an acceptable intermediary step toward functional cure because partial cure is more achievable in the short term, has been shown to lead to a reduction in clinical outcomes,1 and could expedite drug development.
Natural history of chronic HBV infection
The natural history of chronic HBV infection is variable and dependent on a complex interplay between the host immune response and the virus. Chronic HBV infection comprises four phases defined by three clinical parameters: serum alanine aminotransferase (ALT) concentrations, serum HBV DNA levels, and hepatitis B e antigen (HBeAg) status (Fig. 1). The first phase is characterised by the presence of HBeAg and high serum HBV DNA but normal ALT levels. It has been called the "immune-tolerant" phase, though recent studies have challenged the concept of immune tolerance. HBV-specific T-cell responses have been observed in patients in the immune-tolerant phase with similar frequency as in patients in the immune-active phase. This finding has led some to propose that the immune response is better characterised as low inflammatory during the immune-tolerant phase (as opposed to inflammatory during the immune-active phase).[2], [3], [4] The immune-tolerant phase is followed by the "HBeAg-positive immune-active" phase, when ALT levels become elevated. After varying intervals, seroconversion from HBeAg to hepatitis B e antibody occurs, and a majority of patients transition to the "inactive carrier" phase, during which ALT levels return to normal and serum HBV DNA levels are low or undetectable. In some patients, serum HBV DNA and ALT levels become elevated again, after years or decades. These patients are considered to be in the "HBeAg-negative immune-active" phase, which is characterised by fluctuating HBV DNA and ALT levels. The annual incidence of HBeAg-negative immune-active hepatitis among inactive carriers is estimated to be 0.37%.5 Some patients do not fit into any of these conventional phases. Serum HBsAg levels are highest during the immune-tolerant phase and lowest during the inactive carrier phase. Consequently, quantification of serum HBsAg levels may help in determining the phase of infection, particularly for HBeAg-negative persons,6 and to predict the risk of disease progression and HCC in HBeAg-negative patients with low viraemia (HBV DNA <2,000 IU/ml).7 Because phases of chronic HBV infection are defined based on clinical and not immunologic measures, the recent European Association for the Study of Liver Diseases proposed to describe these four phases as phases 1, 2, 3, and 4.8 Patients in phases 1 and 2 would be HBeAg-positive, those in phases 3 and 4 would be HBeAg-negative, those with inactive disease (phases 1 and 3) would be considered to have chronic infection, while those with active disease (phases 2 and 4) would be considered to have chronic hepatitis.
Some patients spontaneously clear HBsAg, but this event is rare, occurring at a rate of 0.5%-1% per year. These patients remain positive for hepatitis B core antibody, and some may develop anti-HBs. The majority of patients who clear HBsAg have undetectable HBV DNA in serum, but HBV DNA persists in the serum in some and in the liver in all patients. These patients are considered to have occult HBV infection. While the risk of cirrhosis and end-stage liver disease is greatly diminished, the risk of HCC after HBsAg loss remains substantial, particularly if HBsAg loss occurred after the age of 50 or after development of cirrhosis.[9], [10], [11] Importantly, HBV can be reactivated upon immunosuppression, suggesting that eradication of HBV from the host is rarely achieved.12
Identifying individuals at greatest risk for development of cirrhosis and HCC is an important goal in the management of chronic HBV infection. Recent studies have highlighted the importance of viral load in predicting risk of cirrhosis and HCC.[13], [14] However, many other host (sex, age, family history of HCC, obesity, diabetes), viral (HBV genotype and variants, coinfection with other viruses: hepatitis C, hepatitis D, human immunodeficiency virus), and environmental (alcohol, smoking, carcinogens) factors contribute to liver disease progression.
Several risk models have been developed to predict risk of HCC.[15], [16], [17], [18], [19] Most of these models were derived from data on Asians. The applicability of these models to all racial/ethnic groups and HBV genotypes, patients with or without cirrhosis, and untreated patients as well as those receiving antiviral therapy has not been confirmed. Cirrhosis is the major risk factor for HCC. In the past decade, noninvasive assessment-serum marker panels and liver stiffness measurements-have largely replaced liver biopsies in staging of liver fibrosis. These noninvasive tests have also been shown to predict survival and HCC in patients with chronic HBV infection because they have high accuracy in diagnosing cirrhosis.[20], [21], [22], [23], [24]
Current status of HBV treatment
Goals of treatment and efficacy assessment
The goals of HBV therapy are to prevent the development of cirrhosis, hepatic decompensation, HCC, and death from HBV-related liver disease. The eventual clinical outcomes of chronic hepatitis B require years, if not decades, of surveillance; therefore, biochemical (normalisation of ALT), virological (suppression of serum HBV DNA), serological (HBeAg and/or HBsAg loss with or without seroconversion to hepatitis B e antibody and anti-HBs), and histological (decrease in hepatic necroinflammation with or without improvement in fibrosis) markers have been used as surrogates for clinical outcomes-cirrhosis, hepatic decompensation, HCC, and HBV-related mortality-and to assess indications for treatment, response, and prognosis. The durability of responses after treatment discontinuation is variable.
Indications for treatment
The decision to treat is based upon clinical assessment of the risk of disease progression, which is related to phase of disease. This risk assessment is based primarily on HBV DNA and ALT levels and the stage of disease, as assessed by liver biopsy or noninvasive staging of hepatic fibrosis. Current guidelines recommend treatment for patients with cirrhosis or decompensated liver disease and, for patients without cirrhosis, evidence of modest to high viraemia and biochemical or histological evidence of hepatic necroinflammation.[25], [26], [27] Continued high levels of HBV replication together with hepatic inflammation increases the risk of cirrhosis and HCC; however, currently available therapies have lower efficacy in patients in the immune-tolerant phase, and treatment is not recommended for these patients.[28], [29]
Approved therapies
Two classes of antiviral therapies have been approved for treatment of hepatitis B: IFNs and NAs. An advantage of IFN is that it results in higher rates of HBeAg and HBsAg loss (particularly in patients with genotype A infection) compared to NAs. Pegylated IFN administered for 48-52 weeks results in HBeAg seroconversion in 24%-27% and HBsAg loss in 3%-7% of patients compared to 12%-22% HBeAg loss and 0%-3% HBsAg loss after the same duration of NA therapy.30 Response to IFN is also more durable, and HBeAg and HBsAg loss may occur after cessation of treatment, while virological relapse, even after HBV DNA has become undetectable, is frequent after cessation of NA.31 However, IFN is less effective at suppressing viral replication compared to NAs, requires parenteral administration, is associated with numerous adverse effects, and is contraindicated in patients with decompensated cirrhosis or severe exacerbations of hepatitis and those with autoimmune or psychiatric illnesses. NAs are administered orally and have negligible adverse effects. The recommended first-line NAs, entecavir and tenofovir, have low risk of drug resistance; but the requirement for indefinite therapy in the majority increases the cost and the risk of nonadherence and adverse effects.
Various combinations of IFN and NA have been evaluated, but most studies have not shown an added benefit compared to monotherapy. A recent study showed that combination of pegylated IFN and tenofovir increased the rate of HBsAg loss to 9% at week 72, but the benefit was mainly observed in patients with genotype A.32 Therefore, there is an urgent need to develop new therapies for HBV that can result in durable suppression of HBV replication and an ensuing decrease in hepatic inflammation and fibrosis after a finite course of therapy. In addition, more research is needed to identify which patients can safely stop therapy. Importantly HBsAg loss at an older age and after the development of cirrhosis does not eliminate the risk of HCC; however, the benefit of further treatment for preventing HCC has not been proven.33 Treatment that can result in a functional cure at an earlier stage of disease might have a greater impact in preventing HCC.
Lack of impact on NAs on cccDNA
A major hurdle to HBV "cure" is the presence of cccDNA in the hepatocyte nucleus in a nonintegrated form or episome. cccDNA serves as the template for transcription of all viral RNAs including pregenomic RNA (pgRNA) and thus plays a key role in the viral life cycle. There are two sources of cccDNA: incoming virions and recycling of encapsidated DNA from the hepatocyte cytoplasm. The half-life of cccDNA is long, thus explaining why it is difficult to cure HBV infection and why HBV can reactivate either spontaneously or following immune suppression, many years after clearance of HBsAg. Chain terminating NAs block the reverse transcription of pgRNA to HBV DNA, but they have a marginal effect on cccDNA production, stability, or transcription. Continued transcription from cccDNA and integrated viral genomes may explain the relatively minor decrease in serum HBsAg levels during NA therapy despite undetectable serum HBV DNA levels.34 Unfortunately, current assays for circulating HBsAg cannot distinguish the transcription of HBsAg from cccDNA vs. integrated HBV DNA.
Impact of antiviral treatment on clinical outcomes
Most, but not all, long-term follow-up studies and meta-analyses indicate that IFN and NA treatment decrease the risk of HCC and liver-related mortality.35A landmark randomised controlled trial showed that the first-generation NA lamivudine decreased the risk of disease progression and HCC in patients with advanced fibrosis or cirrhosis and high viraemia.1 Several studies have shown that maintained viral suppression during NA therapy is associated with regression of fibrosis, reversal of cirrhosis, and reduction in rates of hepatic decompensation.[36], [37] The risk of HCC is also diminished, although not eliminated, making it the only major complication during NA treatment. The observed reduction in the incidence of HCC appears to be more evident in persons with cirrhosis and after several years of continued treatment.35
Novel antiviral therapies
Recent major scientific discoveries have allowed a better understanding of the HBV life cycle, including identification of the cellular receptor for HBV entry, information on the key nuclear enzymes involved in cccDNA formation and on its epigenetic control, the observation of partial cccDNA degradation induced by IFN or nuclear factor-κB signaling pathways, and identification of the role of the hepatitis B x protein (HBx) in HBV transcription. Improved cellular and animal models have also enhanced the in vitro and in vivo assessment of the antiviral activity of novel compounds and their potential toxicity. These major advances in hepatitis B basic research have paved the way for the identification of multiple new therapeutic targets (Fig. 2), essential progress toward a "cure" for hepatitis B. A list of novel antiviral therapies tested in clinical trials is provided in Table S1.
Fig. 2
HBV life cycle and antiviral targets. HBV entry: Lipopeptides mimicking pre-S1 domain competing with Dane particle for binding to NTCP (e.g., Myrcludex B). Other small molecule inhibitors are in development. Targeting cccDNA: Prevention of cccDNA formation. Damage and destruction through cytokines or cccDNA sequence-specific nucleases. Functional silencing through modulation of host cellular epigenetic-modifying enzymes by cytokines or inhibition of viral protein function. HBV mRNAs: siRNA approaches or antisense oligonucleotides to block viral replication and viral protein expression. HBV polymerase: Reverse transcriptase inhibitors include approved NAs. Ribonuclease H inhibitors are in preclinical evaluation. Nucleocapsid assembly and pgRNA packaging: Capsid assembly modulators can affect nucleocapsid assembly and pgRNA encapsidation and may affect the nuclear functions of hepatitis B core protein (cccDNA regulation and IFN-stimulated gene expression). Targeting HBsAg: Phosphorothioate oligonucleotides inhibiting HBsAg release and monoclonal antibodies to decrease circulating HBsAg load are under evaluation. cccDNA, covalently closed circular DNA; dslDNA, double-strand linear DNA; IFN, interferon; HBV, hepatitis B virus; MVB, multivesicular body; NTCP, sodium taurocholate cotransporting; pgRNA, pregenomic RNA; rcDNA, relaxed circular DNA.
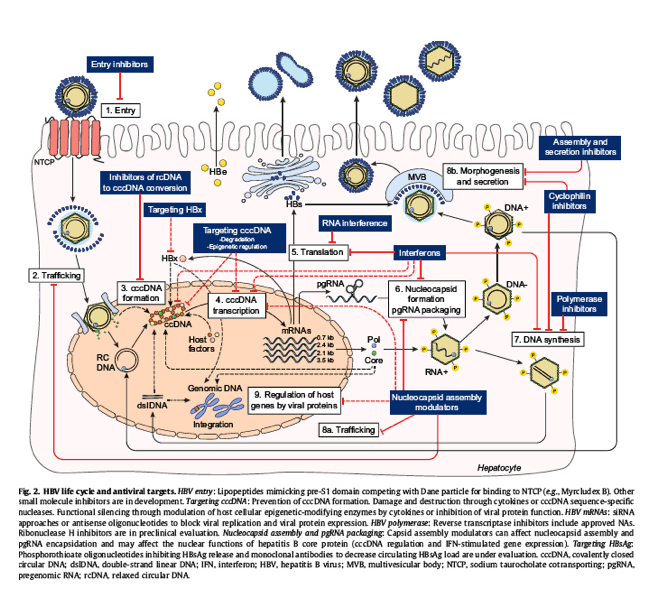
HBV entry inhibitors
The pathway by which HBV enters hepatocytes involves virus attachment to heparan sulfate proteoglycans, allowing the binding of HBV pre-S1 to human sodium taurocholate cotransporting polypeptide (NTCP), followed by membrane fusion and nucleocapsid release in the cytoplasm of infected cells.
Entry inhibitors can be classified according to their modes of action. (i) Neutralising antibodies target the antigenic loop of HBV S-domain or N-terminal epitopes in the pre-S1 domain. These antibodies are highly specific but require parenteral administration, and large quantities of antibodies are necessary to neutralise circulating subviral envelope particles. (ii) Attachment inhibitors are negatively or positively charged drugs that bind the virus (e.g., heparin) or cellular heparan sulfate proteoglycans (e.g., poly-l-lysine). They are efficient but not specific. (iii) Substrates of NTCP include conjugated bile salts (e.g., taurocholate, ezetimibe) and other small molecules that are transported by NTCP; the requirement for high concentrations and short half-time at the receptor limit their clinical application. (iv) Irreversible NTCP inhibitors: myrcludex B (myristoylated pre-S1 peptide), cyclosporin A, and derivatives are allosteric inhibitors of NTCP. They irreversibly block receptor function at nonsaturating concentrations and have long half-time at the receptor, but they can block transport of bile salts and other NTCP substrates at higher concentrations.
Entry inhibitors may prevent de novo cccDNA formation in noninfected hepatocytes and might be more effective at preventing mother-to-child transmission or reinfection after liver transplantation than at eliminating HBV in chronically infected persons.
Clinical trials of myrcludex B with or without pegylated IFN in chronic HBV and chronic hepatitis D virus infection are under way.[38], [39]
Targeting cccDNA
Damage and destruction
A number of studies demonstrate that it might be possible to target cccDNA despite its location in the protected sanctuary of the hepatocyte nucleus. Studies in cultured hepatocytes showed that several cytokines (IFN-α, lymphotoxin-β receptor agonists, IFN-γ, and tumour necrosis factor-α) can modulate pathways leading to the up-regulation of APOBEC3A/B deaminases, which in turn induced nonhepatotoxic degradation of cccDNA. However, only partial degradation was achieved in these tissue culture studies.40 DNA cleavage enzymes, including homing endonucleases or meganucleases, zinc-finger nucleases, TAL effector nucleases, and CRISPR-associated (cas) nucleases, specifically targeting cccDNA are being explored in experimental models.41Some studies suggest that editing of HBV DNA by CRISPR-associated (cas) nuclease cleavage is more efficient than APOBEC-mediated cytosine deamination following treatment of infected cells with IFN.42 Efficient delivery of these gene editing approaches to HBV-infected hepatocytes without unintended off-target effects will need to be addressed before they can be tested in clinical trials.
Functional silencing
Viral cccDNA is organised into a chromatin-like structure and is subject to epigenetic regulation.43 Epigenome modifiers can potentially block cccDNA transcriptional activity and shut down viral protein expression; however, if not specific, they may induce harmful effects on host cells. Some cytokines, including IFN-α, have been shown to decrease cccDNA transcription through epigenetic modifications in preclinical models.44 HBx has been shown to be required for cccDNA transcription and viral replication through degradation of the Smc5/6 restriction factor; thus, HBx may be an attractive viral target to silence not only cccDNA transcription45 but also several other HBx-dependent, virus-related cellular interactions. Current assays to detect cccDNA lack specificity, and a standardised method to measure cccDNA or cccDNA transcription is needed.
Targeting viral transcripts
The use of RNA interference to inhibit replication of HBV has been extensively evaluated in vitro and validated in animal models. Small interfering RNA (siRNA) can be designed to target any viral transcript and induce its degradation by the RISC/Ago2 complex, resulting in gene silencing. The potential limitations are the need for intravenous administration, the risk of off-target binding, the potential toxicity of the vehicle, and the risk of immune activation by pattern recognition receptors. Three siRNA formulations with different modes of delivery are under preclinical evaluation and/or early-phase clinical trial. Preliminary results of a phase II trial showed that a single dose of ARC-520 in combination with entecavir resulted in a profound and durable decrease in serum HBV DNA in both HBeAg-positive and HBeAg-negative patients and a decrease in HBsAg level in HBeAg-positive, but not in HBeAg-negative, patients.[46], [47] The siRNAs were designed to target the cotermini of all transcripts from cccDNA, and the lack of effect on HBsAg level in HBeAg-negative patients was postulated to be due to altered viral transcript sequences derived from integrated HBV DNA. Further studies are needed to determine whether the decline in HBsAg production alone might be sufficient to restore HBV-specific T-cell response or if addition of other antiviral or immune modulatory therapies is needed.
Antisense oligonucleotides targeting viral transcripts can block viral protein expression through steric blockade of protein translation and/or RNA degradation by ribonuclease H cleavage. In vitro and in vivo preclinical evaluations have shown their potential to inhibit viral replication and decrease viral antigen load, but optimal delivery remains a challenge.
Nucleocapsid assembly and pgRNA packaging
Nucleocapsid formation and packaging of the pgRNA (the template for viral replication) are critical steps of the viral life cycle. Therefore, developing inhibitors or modulators of this process is an attractive therapeutic approach. The HBV core protein is involved in many aspects of the viral life cycle including transport of viral genome to the nucleus, uncoating to release relaxed circular DNA in the nucleus, packaging of polymerase protein and pgRNA, capsid assembly, modulation of reverse transcription, and interaction with envelope proteins for virus assembly. It may have additional functions including modulation of cccDNA chromatin and stability, nuclear export of viral RNAs, and modulation of innate immunity.
The HBV precore/core protein has recently emerged as a promising direct antiviral target. With the knowledge of the three-dimensional structure of the core protein, several classes of non-nucleoside small molecules called core protein assembly modulators have been developed, including phenylpropenamide and heteroaryldihydropyrimidine derivatives. These molecules can strengthen protein-protein interaction, inhibit pgRNA encapsidation, and block plus strand DNA synthesis.[48], [49] Results of the first dose-ranging phase Ib study of NVR3-778 showed a decline in serum HBV DNA, HBV RNA, and HBsAg, with more pronounced effect when combined with pegylated IFN.50
Targeting HBsAg
Strategies inhibiting viral gene expression either through cccDNA transcription or viral mRNA translation can decrease serum HBsAg levels. Owing to the large amount of circulating HBsAg in persons with chronic HBV infection, monoclonal antibodies aimed at neutralising and/or depleting HBsAg from the bloodstream are unlikely to be effective unless used in combination with other approaches.
Nucleic acid polymers have been shown to decrease secreted HBsAg, through as yet unknown mechanisms. Clinical trials of REP 2055 and REP 2139 used in monotherapy followed by combination with pegylated IFN or thymosin alpha-1 induced a marked decline in circulating HBsAg levels and viraemia and anti-HBs seroconversion in some patients.51 Similar results were observed in pilot studies of tenofovir and pegylated IFN in combination with REP 2139 or REP 2165.52 These results need to be replicated in larger studies, and the potential for cytotoxicity resulting from intracellular retention of HBsAg must be resolved.
Immune response to HBV and Implications for immune modulatory therapies
An orchestrated innate and adaptive immune response is necessary to sense and control HBV infection. The induction of innate proinflammatory responses and cytokine activation are weak in chronic HBV infection.53 IFN-γ production from natural killer cells is detectable in acute hepatitis but deficient in chronic hepatitis. HBV-specific T cells necessary for HBV control (which may also play a role in hepatic inflammation) are dysfunctional and may be depleted or deleted in chronic HBV.[54], [55] Their exhausted phenotype has been attributed to persistent antigen exposure and increased expression of T-cell inhibitory responses,[3], [53], [56] providing the basis for the focus on reducing HBsAg production. B-cell dysfunction in chronic hepatitis B is less well characterised, though the high rate of HBV reactivation in patients receiving anti-cluster of differentiation 20 suggests that B cells play a role in immune control of chronic HBV. Accordingly, several potential target mechanisms for immune modulation to engender or restore HBV-specific immune responses in conjunction with profound inhibition of HBV replication and HBsAg production to attain immunological control have been suggested (Fig. 3). The main concerns of immune modulatory therapy are the induction of uncontrolled hepatitis flares and autoimmunity.
Interferons
IFN-α has been used to treat chronic HBV and chronic hepatitis D virus. IFN-α induces expression of IFN-sensitive genes encoding intracellular or secreted effector proteins with antiviral properties. Recent studies have shown that IFN-α also inhibits pgRNA encapsidation, enhances cccDNA degradation, and exerts epigenetic modification of cccDNA transcription.[40], [57] Response to IFN-α (HBeAg seroconversion) is durable in >70% of patients.[58], [59]Understanding the mechanisms for IFN nonresponsiveness and the higher rates of response in genotype A infection could lead to new targets for antiviral and/or immune modulatory therapies.
Pathogen recognition receptors
Pathogen recognition receptors are an important gateway to sensing by the innate immune system. Pharmacological activation of the intrahepatic innate immune response with toll-like receptor 7, 8, or 9 has been studied. GS-9620, an oral agonist of toll-like receptor 7, induced a decline in serum HBV DNA and HBsAg levels as well as hepatic HBV DNA in HBV-infected chimpanzees.60 Similar effects were also observed in woodchucks61 but not in humans.[62], [63] The discrepancies between results in animal models and humans highlight the importance of testing new therapies in humans at an early stage in drug development.
Stimulator of IFN gene agonists
Stimulator of IFN gene is the adapter protein of multiple cytoplasmic DNA receptors and a pathogen recognition receptor recognising bacterial second messenger and may be a potential target of pharmacologic activation of the innate immune response.64 Stimulator of interferon gene agonists can also be used as adjuvants to therapeutic vaccination. Retinoic acid-inducible protein 1 has been shown not only to induce IFN and cytokine production but also to inhibit HBV replication through sensing of the epsilon structure of pgRNA.65 SB 9200, an oral prodrug of the dinucleotide SB 9000, is thought to activate retinoic acid-inducible protein 1 and nucleotide-binding oligomerisation domain-containing protein 2, resulting in IFN-mediated antiviral immune responses in virus-infected cells and decreased serum woodchuck hepatitis virus DNA and surface antigen levels.66 Clinical trials are ongoing.
Checkpoint modulation
The lack of a T cell-mediated response in chronic HBV is partly due to overexpression of coinhibitory receptors including programmed cell death, cytotoxic T lymphocyte-associated antigen-4 lymphocyte activation gene, and mucin domain to impair T-cell effector function. T-cell function may also be impaired by immunosuppressive cytokines including interleukin 10. Recent cancer therapies have indicated the potential of blockade of these coinhibitory receptors with antibodies. Such inhibition may reverse immune dysfunction in hepatitis B as demonstrated in studies in woodchucks and ex vivo studies in humans.[4], [67], [68], [69] The main concerns of this approach are induction of uncontrolled hepatitis flares and autoimmunity, which can lead to fatal organ damage.70
Therapeutic vaccine
The goal of therapeutic vaccination is to stimulate or boost the host immune response to restore immune control, resulting in sustained suppression of HBV replication and ultimately HBsAg loss. Therapeutic vaccination strategies including conventional HBsAg vaccine with or without potent adjuvants, T-cell vaccine, immune complexes of HBsAg and human anti-HBs, apoptotic cells that express HBV antigens, DNA vaccines, and viral vectors expressing HBV proteins have been evaluated with limited success. Patients with high viral loads may be less responsive, and patients with cirrhosis may be at higher risk for immune-mediated hepatitis flares. Pretreatment with NAs to suppress HBV replication may enhance HBV-specific T-cell response and prevent flares. GS-4774, a heat-inactivated yeast-based vaccine expressing HBV S/C/X fusion protein, induced HBV-specific T-cell responses in healthy volunteers; but HBsAg decline and HBsAg loss were not observed in chronic hepatitis B patients virally suppressed on NAs as well as those who were NA-naive.[71], [72], [73] A list of therapeutic vaccines tested in clinical trials is provided in Table S1.
Combination of antiviral and immune modulatory therapies
Combinations of antiviral therapy targeting multiple steps in the HBV life cycle to suppress viral replication and viral antigen production and immune modulatory therapy to restore immune response to HBV will likely be needed to achieve the goal of HBV "cure," but which specific combination of agents will be needed is not certain because most antiviral or immune modulatory drugs in clinical evaluation are entering either phase Ib or phase II trials at this time. The possibility that HBV-specific T-cell function can be partially restored has been demonstrated in patients with chronic HBV who had spontaneous or antivirus (IFN or NA)-mediated HBeAg or HBsAg loss.[74], [75] With the availability of novel approaches such as siRNA or combination of antiviral agents targeting different steps in the HBV life cycle, marked reduction and even shutdown of HBsAg production may be achieved in a higher proportion of patients. The reduction in antigen load might facilitate the restoration of HBV-specific T-cell responses. Historically, immune modulatory therapies have focused on patients in the immune-active phase, but recent studies suggest that patients in the "immune-tolerant or high-replication noninflammatory" phase should also be considered as HBV-specific T cells are present in this phase of disease.[3], [76] The ability to "cure" hepatitis B at earlier stages of liver disease would theoretically have a greater impact on reducing risk of HCC.
Clinical trials aimed at HBV cure
Efficacy endpoints for clinical trials
Initial trials of IFN and NA used biochemical, virological, serological, and histological endpoints to assess efficacy, while more recent trials have focused on virological and serological endpoints because these endpoints have been shown to correlate with improved clinical outcomes.[36], [77], [78], [79] The use of a biochemical endpoint is problematic because of the lack of a standardised definition of normal ALT. Furthermore, with increasing prevalence of obesity and non-alcoholic fatty liver disease, failure to normalise ALT may not necessarily indicate ongoing liver inflammation induced by HBV. Indeed, phase III clinical trials of NAs have consistently found a lower percent of patients achieving a biochemical vs. a virological response. Noninvasive assessments of liver fibrosis have replaced liver biopsies in assessing liver disease in clinical practice, and a histological endpoint would no longer be practical or necessary for assessing functional cure. However, liver biopsies may be required in proof-of-concept studies to confirm a novel mode of action and/or to validate noninvasive surrogate markers of antiviral activity.
For both antiviral and immune modulatory therapy trials, survey respondents ranked suppression of serum HBV DNA to undetectable and loss of HBsAg as the most important primary efficacy endpoint for phase II and phase III trials, respectively; and the expert panel agreed. The difference in ranking of primary efficacy endpoint for phase II vs. phase III trials reflects a desire to have an earlier readout in phase II trials and the potential difficulty in achieving HBsAg loss after shorter treatment. HBV DNA suppression will not be an appropriate endpoint for trials enrolling patients who are virally suppressed on NAs. Decline in serum HBsAg level has been used as an endpoint in some trials, but there is no consensus on whether the kinetics of decline in HBsAg level can predict ultimate HBsAg loss.
There was general consensus (∼65%) that the most appropriate time to assess the primary efficacy endpoint for phase III trials of novel antiviral or immune modulatory therapies should be month 6 posttreatment. The choice reflected the intent to achieve a durable response ("cure") after a finite course of treatment. There was less consensus on the optimal time for assessing efficacy in phase II trials, with the responses divided between month 6 on treatment vs. month 6 posttreatment. The expert panel indicated that the most appropriate time to assess efficacy endpoints may depend on the mechanism of action and half-life of the drug. For example, drugs that inhibit reverse transcription of HBV pgRNA to HBV DNA are expected to result in rapid decline in serum HBV DNA levels, while drugs aimed at restoring immune response may take longer to have any measurable effects on viral load. The expert panel also emphasised the need for long-term follow-up to confirm durability of responses and impact on clinical outcomes.
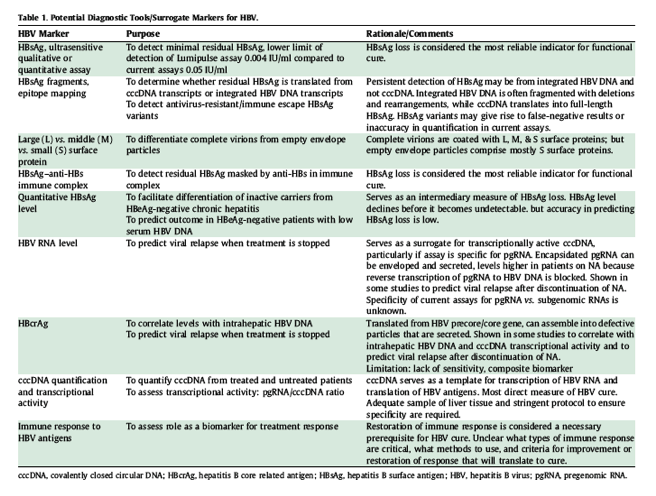
Diagnostic markers for new assays to determine therapeutic efficacy
There was consensus on the need for standardised assays to provide mechanistic insights into the effects of novel antiviral or immune modulatory agents and to have new surrogate markers to assess HBV "cure" (Table 1).
Serum HBsAg assays
HBsAg loss was ranked as the most important efficacy endpoint for phase III trials of both novel antiviral and immune modulatory therapies. Some experts suggested the need for more sensitive HBsAg assays. Others indicated that the persistence of low levels of HBsAg in serum may stem not from persistent cccDNA transcription but from integrated HBV DNA genomes. Assays that can differentiate HBsAg originating from integrated HBV DNA vs. cccDNA are required. Failure to detect HBsAg in serum may not represent shutdown of HBsAg production as circulating HBsAg may be complexed to anti-HBs. HBV produces a surplus of empty envelope particles, which can outnumber virions by >1,000-fold. Assays that can differentiate surface proteins in virions vs. subviral particles (large, middle vs. small HBsAg proteins) would be informative.
Serum HBsAg decline may be used as a surrogate to explore efficacy in early-phase trials, although its accuracy in predicting HBsAg loss has not been established. Quantitative HBsAg assays can also be used to categorise disease stage and to prognosticate. Several standardised quantitative HBsAg assays have been available in Europe and in Asia in the last decade, and the consensus is that they will be essential tools for new drug development.
cccDNA quantification and transcriptional activity
Many new antiviral agents in development aim to enhance elimination or degradation of cccDNA or to epigenetically modify cccDNA transcription. Assays that reliably measure concentrations of cccDNA and/or its transcriptional activity would directly assess efficacy of these agents and would be invaluable in proof-of-concept studies. Currently, there are no standardised assays for intrahepatic cccDNA. However, there was consensus that an adequate sample of liver tissue would be necessary and that appropriate measures to ensure specificity are needed, particularly in the presence of abundant relaxed circular DNA.
Assays for serum markers that are reliable surrogates of intrahepatic cccDNA would be desirable for clinical trials. Several studies have shown that serum HBsAg level correlates better with hepatic cccDNA than serum HBV DNA level, suggesting that it is an indirect marker of cccDNA.[80], [81] However, while the correlation is satisfactory in HBeAg-positive patients, it is suboptimal in HBeAg-negative patients.[80], [82] HBsAg can be translated from subviral RNAs transcribed from cccDNA or from integrated HBV DNA. It has been suggested that HBsAg is predominantly derived from cccDNA in HBeAg-positive patients, while an increasing proportion is derived from integrated HBV DNA in HBeAg-negative persons. Recent studies suggest that serum HBV RNA and hepatitis B core-related antigen (HBcrAg) levels may be more reliable surrogates of hepatic cccDNA than HBsAg levels.
Serum HBV RNA assays
The vast majority of circulating HBV virions contain partially double-stranded relaxed circular DNA; however, circulating "virions" containing HBV RNA had been reported.83 These RNA-containing "virions" are more abundant in patients receiving NA therapy, probably because inhibition of reverse transcription of pgRNA by NA leads to accumulation of encapsidated pgRNA, some of which may be enveloped and secreted. Thus, the detection of HBV RNA in serum in the absence of detectable serum HBV DNA in patients receiving NA therapy could infer ongoing cccDNA transcription and has been shown to be a predictor of viral relapse after discontinuation of NA therapy.84 Assays for serum HBV RNA levels, particularly if they are specific for pgRNA, may provide a useful surrogate for transcriptionally active cccDNA.
Serum HBcrAg assays
The HBV precore/core gene is translated into the core protein, precore/core precursor, and HBeAg; these proteins can be collectively measured. HBcrAg can assemble into defective, enveloped particles that that do not contain HBV RNA or HBV DNA. A core-related antigen immunoassay measuring all these forms of precore/core gene expression has been developed. Serum HBcrAg levels have been shown to correlate with hepatic HBV DNA and to predict viral relapse after discontinuation of NA therapy.85 Assays for serum HBcrAg levels may provide indirect evidence for transcriptionally active cccDNA.86
Immune response to HBV
Restoration of immune response to HBV is a key step toward HBV "cure." Most immune modulatory therapies have focused on restoring T-cell response. While markers of response to antiviral therapies can be used to evaluate immune modulatory therapies, standardised assays to measure improvement in specific immune responses to predict viral clearance would be informative in early-phase drug development. The development of standardised assays to assess restoration of immune responses to HBV will require consensus on the requisite assays; their methodology, sensitivity, and specificity; their correct interpretation and reproducibility in different laboratories; and cross-validation of the assays.
Approval of HBV diagnostic assays
Development of standardised assays for surrogate endpoints for HBV cure should occur in parallel with development of novel antiviral and immune modulatory therapies to expedite research. During the closed session, the US Food and Drug Administration and European Medicines Agency representatives proposed consideration of simultaneous testing of multiple intermediate secondary endpoints in all phase II and III trials. The research assays must be standardised and data and assay platforms shared to facilitate identification of the appropriate surrogate markers of efficacy. It is important that the approval of new treatments is linked to the development and approval of new diagnostic assays used to measure efficacy or to predict response.
Assessment of safety and stop rules
The remarkable safety profile of current NAs imposes a stringent requirement for the safety of new HBV therapies at all stages of the disease. A unique concern in the development of hepatitis B therapies is the risk of severe hepatitis flares, which can result in hepatic decompensation and death. The US Food and Drug Administration has explicit recommendations on managing drug-induced liver injury; however, these recommendations do not apply to patients with underlying liver disease. Furthermore, transient hepatitis flares are not always harmful and may portend immune clearance of infected hepatocytes. Hepatitis flares may be due to direct drug-induced liver injury, drug-induced immune-mediated hepatitis (e.g., checkpoint inhibitors), the underlying disease (incomplete viral suppression), or immune clearance of infected hepatocytes (successful viral suppression and accompanying restitution of the host immune response). The timing and course of the flare, as well as the associated chronological changes in serum HBV DNA levels, can help in differentiating the cause of most, but not all, hepatitis flares; for example, flares related to drug-induced liver injury may be idiosyncratic and occur at any time. There was no consensus on definition of hepatitis flares (magnitude of ALT increase, absolute value, or fold-change compared to baseline); however, a majority of survey respondents and the expert panel agreed that flares accompanied by increase in bilirubin or prothrombin time and flares in patients with cirrhosis should be considered severe. Other adverse events can also occur, e.g., off-target effects of siRNA or epigenetic modification, autoimmunity associated with checkpoint inhibitors, and hepatotoxicity from retention of dysfunctional viral particles. There was no consensus on when a trial or development of a new agent should be stopped due to safety concerns; however, survey respondents and the expert panel indicated that any death or liver transplantation, hepatic decompensation, irreversible autoimmunity, or incidence of severe hepatitis flare in >5% of patients could prompt a halt. Two-thirds of respondents to the survey and most of the expert panel recommended that safety and efficacy should be assessed for at least 6 months after stopping therapy.
Design of clinical trials for HBV cure
A combination of antiviral and immune modulatory therapies will most likely be needed to increase the likelihood of HBV cures. There was consensus that the antiviral activity and safety of individual new agents used as monotherapy and infrequent or insignificant drug interactions should be first established before progressing to clinical trials of combination therapy. However, demonstration of efficacy as a monotherapy need not be required.
New therapies are most needed for patients at high risk of HBV-associated mortality (cirrhosis) and those in whom current therapies are less effective (immune-tolerant, hepatitis D virus infection). However, in designing early-phase clinical trials, the target patient populations will necessarily be those who are most likely to respond and able to tolerate possible exacerbations of hepatitis. The consensus was to initially conduct trials in treatment-naïve, HBeAg-positive patients with active disease or in HBeAg-positive or HBeAg-negative patients virally suppressed on NAs. Proof-of-principle and safety data would first be established in patients without cirrhosis.
A particular challenge with designing phase II studies is to identify a target population with sufficient heterogeneity to be representative of the population with chronic hepatitis B but not so diverse that it hinders analysis because of multiple subgroups. Among the survey respondents, 59% felt that patient subpopulations should be studied separately. If multiple patient populations were included in the same trial, patients could be potentially stratified for cirrhosis, HBeAg status, HBsAg level, HBV DNA level, treatment history, and HBV genotype. Standardised criteria for ascertaining cirrhosis by noninvasive serum markers and/or liver elastography are required.
Given the heterogeneity of the natural course of chronic hepatitis B, there was consensus that randomised controlled trials are needed to establish efficacy and that comparison with placebo is feasible and ethical in trials for patients in the immune-tolerant or inactive phases because current guidelines do not recommend treatment for these patients. For patients with active disease or cirrhosis, investigational new agents can be compared to NA or IFN. Alternatively, the new drug can be tested against placebo as an additional therapy to NA. There was consensus that the trials should aim to demonstrate superiority of the investigational therapy.
Summary and recommendations
A summary of the recommendations on endpoints and design of clinical trials for HBV cure is presented in Table 2. Improved understanding of the HBV life cycle and host immune response to HBV has facilitated discovery and design of antiviral therapies directed against multiple steps of the HBV replication cycle, as well as potential immunomodulatory therapies to restore immune response to HBV infection. Development of novel HBV therapies will be further aided by the availability of improved cellular and animal infection models. While this progress has raised the possibility of a cure for hepatitis B, a complete sterilising cure, i.e., viral eradication from the host, may be unrealistic due to the presence of integrated HBV DNA. Although HBsAg clearance is infrequent, it does occur with NA and IFN therapy and is associated with improved clinical outcomes. Therefore, a functional cure (characterised by sustained loss of HBsAg with or without anti-HBs seroconversion) after a finite course of novel antiviral and immune modulatory therapies in a higher proportion of patients than is currently achieved with existing treatments is an attainable goal.
Cooperation between academia, industry, and regulatory agencies to standardise and validate surrogate markers for cure, in order to facilitate the development of curative therapies and to facilitate the path from discovery to regulatory approval, is required. Limited proof-of-concept monotherapy studies to evaluate safety and antiviral activity should be conducted prior to proceeding to combination therapy. The safety of any new curative therapies will be paramount given the excellent safety of currently approved NAs. Continued collaboration among the stakeholders will make HBV "cure" a reality for patients with chronic HBV.
.
|
|
| |
| |
|
|
|