| |
FDA - G/P - Trials, Package Insert, MAVYRET approved to treat HCV
|
| |
| |
FDA Mavyret Package Insert - Download the PDF here

Information about FDA HIV product approvals, safety warnings, medical product labeling changes, notices of upcoming public meetings, and notices about proposed regulatory guidances.
On August 3, 2017, FDA approved MAVYRET. MAVYRET is a fixed-dose combination of glecaprevir, a hepatitis C virus (HCV) NS3/4A protease inhibitor, and pibrentasvir, an HCV NS5A inhibitor.
MAVYRET is indicated for the treatment of adult patients with chronic HCV genotype 1, 2, 3, 4, 5 or 6 infection without cirrhosis or with compensated cirrhosis (Child-Pugh A). MAVYRET is also indicated for the treatment of adult patients with HCV genotype 1 infection, who previously have been treated with a regimen containing an HCV NS5A inhibitor or an NS3/4A protease inhibitor (PI), but not both
The safety and efficacy ofMAVYRET were evaluated during clinical trials enrolling approximately 2,300 adults with genotype 1, 2, 3, 4, 5 or 6 HCV infection without cirrhosis or with mild cirrhosis. Results of the trials demonstrated that 92-100 percent of patients who receivedMAVYRET for 8, 12 or 16 weeks duration had no virus detected in the blood 12 weeks after finishing treatment, suggesting that patients' infection had been cured. Highlights from the label are summarized below.
DOSAGE AND ADMINISTRATION
Testing Prior to the Initiation of Therapy
Test all patients for evidence of current or prior HBV infection by measuring hepatitis B surface antigen (HBsAg) and hepatitis B core antibody (anti-HBc) before initiating HCV treatment with MAVYRET.
Recommended Dosage in Adults
MAVYRET is a fixed-dose combination product containing glecaprevir 100 mg and pibrentasvir 40 mg in each tablet.
The recommended oral dosage of MAVYRET is three tablets (total daily dose: glecaprevir 300 mg and pibrentasvir 120 mg) taken once daily with food.
Tables 1 and 2 provide the recommended MAVYRET treatment duration based on the patient population in HCV mono-infected and HCV/HIV-1 co-infected patients with compensated liver disease (with or without cirrhosis) and with or without renal impairment including patients receiving dialysis.
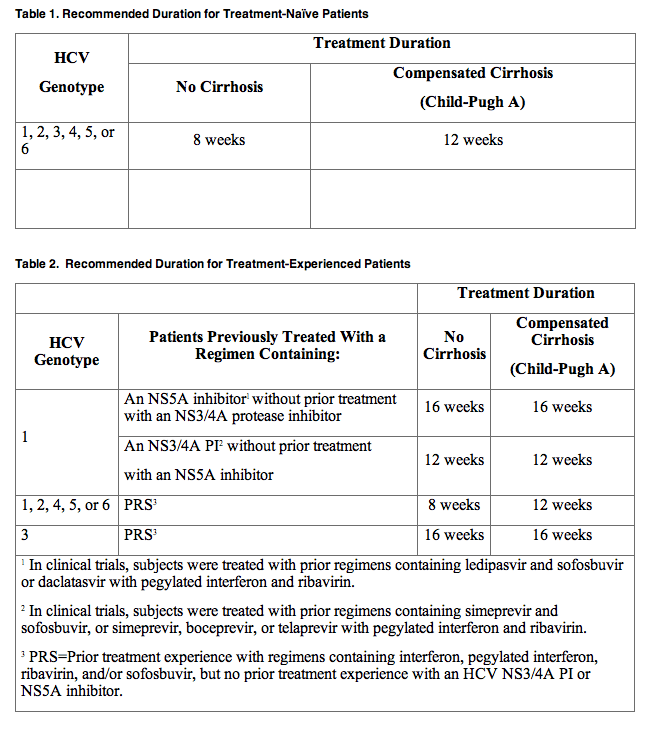
Hepatic Impairment
MAVYRET is not recommended in patients with moderate hepatic impairment (Child-Pugh B) and is contraindicated in patients with severe hepatic impairment (Child-Pugh C)
CONTRAINDICATIONS
MAVYRET is contraindicated in patients with severe hepatic impairment (Child-Pugh C)
MAVYRET is contraindicated with atazanavir or rifampin
WARNINGS AND PRECAUTIONS
Risk of Hepatitis B Virus Reactivation in Patients Coinfected with HCV and HBV
Hepatitis B virus (HBV) reactivation has been reported in HCV/HBV coinfected patients who were undergoing or had completed treatment with HCV direct-acting antivirals, and who were not receiving HBV antiviral therapy. Some cases have resulted in fulminant hepatitis, hepatic failure and death. Cases have been reported in patients who are HBsAg positive and also in patients with serologic evidence of resolved HBV infection (i.e., HBsAg negative and anti-HBc positive). HBV reactivation has also been reported in patients receiving certain immunosuppressant or chemotherapeutic agents; the risk of HBV reactivation associated with treatment with HCV direct-acting antivirals may be increased in these patients.
HBV reactivation is characterized as an abrupt increase in HBV replication manifesting as a rapid increase in serum HBV DNA level. In patients with resolved HBV infection reappearance of HBsAg can occur. Reactivation of HBV replication may be accompanied by hepatitis, i.e., increase in aminotransferase levels and, in severe cases, increases in bilirubin levels, liver failure, and death can occur.
Test all patients for evidence of current or prior HBV infection by measuring HBsAg and anti- HBc before initiating HCV treatment with MAVYRET. In patients with serologic evidence of HBV infection, monitor for clinical and laboratory signs of hepatitis flare or HBV reactivation during HCV treatment with MAVYRET and during post-treatment follow-up. Initiate appropriate patient management for HBV infection as clinically indicated.
Risk of Reduced Therapeutic Effect Due to Concomitant Use of MAVYRET with Carbamazepine, Efavirenz Containing Regimens, or St. John's Wort
Carbamazepine, efavirenz, and St. John's wort may significantly decrease plasma concentrations of glecaprevir and pibrentasvir, leading to reduced therapeutic effect of MAVYRET. The use of these agents with MAVYRET is not recommended
ADVERSE REACTIONS
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of MAVYRET cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Overall Adverse Reactions in HCV-Infected Adults Without Cirrhosis or With Compensated Cirrhosis (Child-Pugh A)
The adverse reactions data for MAVYRET in subjects without cirrhosis or with compensated cirrhosis (Child-Pugh A) were derived from nine Phase 2 and 3 trials which evaluated approximately 2,300 subjects infected with genotype 1, 2, 3, 4, 5, or 6 HCV who received MAVYRET for 8, 12 or 16 weeks.
The overall proportion of subjects who permanently discontinued treatment due to adverse reactions was 0.1% for subjects who received MAVYRET for 8, 12 or 16 weeks.
The most common adverse reactions, all grades, observed in greater than or equal to 5% of subjects receiving 8, 12, or 16 weeks of treatment with MAVYRET were headache (13%), fatigue (11%), and nausea (8%). In subjects receiving MAVYRET who experienced adverse reactions, 80% had an adverse reaction of mild severity (Grade 1). One subject experienced a serious adverse reaction.
Adverse reactions (type and severity) were similar for subjects receiving MAVYRET for 8, 12 or 16 weeks. The type and severity of adverse reactions in subjects with compensated cirrhosis (Child-Pugh A) were comparable to those seen in subjects without cirrhosis.
Adverse Reactions in HCV-Infected Adults treated with MAVYRET in Controlled Trials
ENDURANCE-2
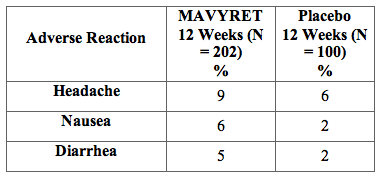
Among 302 treatment-naïve or PRS treatment-experienced, HCV genotype 2 infected adults enrolled in ENDURANCE-2, adverse reactions (all intensity) occurring in at least 5% of subjects treated with MAVYRET for 12 weeks are presented in Table 3. In subjects treated with MAVYRET for 12 weeks, 32% reported an adverse reaction, of which 98% had adverse reactions of mild or moderate severity. No subjects treated with MAVYRET or placebo in ENDURANCE-2 permanently discontinued treatment due to an adverse drug reaction.
Table 3. Adverse Reactions Reported in ≥5% of Treatment-Naïve and PRS-Experienced Adults Without Cirrhosis Receiving MAVYRET for 12 Weeks in ENDURANCE-2
ENDURANCE-3
Among 505 treatment-naïve, HCV genotype 3 infected adults without cirrhosis enrolled in ENDURANCE-3, adverse reactions (all intensity) occurring in at least 5% of subjects treated with MAVYRET for 8 or 12 weeks are presented in Table 4. In subjects treated with MAVYRET, 45% reported an adverse reaction, of which 99% had adverse reactions of mild or moderate severity. The proportion of subjects who permanently discontinued treatment due to adverse reactions was 0%, < 1% and 1% for the MAVYRET 8 week arm, MAVYRET 12 week arm and DCV + SOF arm, respectively.
Table 4. Adverse Reactions Reported in ≥5% of Treatment-Naïve Adults Without Cirrhosis Receiving MAVYRET for 8 Weeks or 12 Weeks in ENDURANCE-3
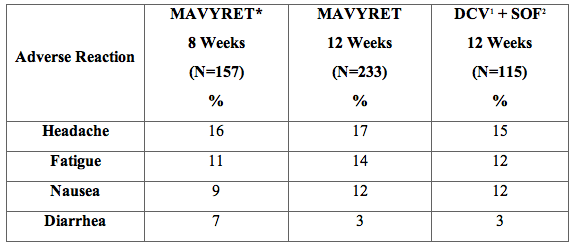
1 DCV=daclatasvir
2 SOF=sofosbuvir
* The 8 week arm was a non-randomized treatment arm.
Adverse Reactions in HCV-Infected Adults with Severe Renal Impairment Including Subjects on Dialysis
The safety of MAVYRET in subjects with chronic kidney disease (Stage 4 or Stage 5 including subjects on dialysis) with genotypes 1, 2, 3, 4, 5 or 6 chronic HCV infection without cirrhosis or with compensated cirrhosis (Child-Pugh A) was assessed in 104 subjects (EXPEDITION-4) who received MAVYRET for 12 weeks. The most common adverse reactions observed in greater than or equal to 5% of subjects receiving 12 weeks of treatment with MAVYRET were pruritus (17%), fatigue (12%), nausea (9%), asthenia (7%), and headache (6%). In subjects treated with MAVYRET who reported an adverse reaction, 90% had adverse reactions of mild or moderate severity (Grade 1 or 2). The proportion of subjects who permanently discontinued treatment due to adverse reactions was 2%.
Laboratory Abnormalities
Serum bilirubin elevations
Elevations of total bilirubin at least 2 times the upper limit of normal occurred in 3.5% of subjects treated with MAVYRET versus 0% in placebo; these elevations were observed in 1.2% of subjects across the Phase 2 and 3 trials. MAVYRET inhibits OATP1B1/3 and is a weak inhibitor of UGT1A1 and may have the potential to impact bilirubin transport and metabolism, including direct and indirect bilirubin. No subjects experienced jaundice and total bilirubin levels decreased after completing MAVYRET.
Drug Interactions
Mechanisms for the Potential Effect of MAVYRET on Other Drugs
Glecaprevir and pibrentasvir are inhibitors of P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), and organic anion transporting polypeptide (OATP) 1B1/3. Coadministration with MAVYRET may increase plasma concentration of drugs that are substrates of P-gp, BCRP, OATP1B1 or OATP1B3. Glecaprevir and pibrentasvir are weak inhibitors of cytochrome P450 (CYP) 3A, CYP1A2, and uridine glucuronosyltransferase (UGT) 1A1.
Mechanisms for the Potential Effect of Other Drugs on MAVYRET
Glecaprevir and pibrentasvir are substrates of P-gp and/or BCRP. Glecaprevir is a substrate of OATP1B1/3. Coadministration of MAVYRET with drugs that inhibit hepatic P-gp, BCRP, or OATP1B1/3 may increase the plasma concentrations of glecaprevir and/or pibrentasvir.
Coadministration of MAVYRET with drugs that induce P-gp/CYP3A may decrease glecaprevir and pibrentasvir plasma concentrations.
Carbamazepine, efavirenz, and St. John's wort may significantly decrease plasma concentrations of glecaprevir and pibrentasvir, leading to reduced therapeutic effect of MAVYRET. The use of these agents with MAVYRET is not recommended Established and Other Potential Drug Interactions
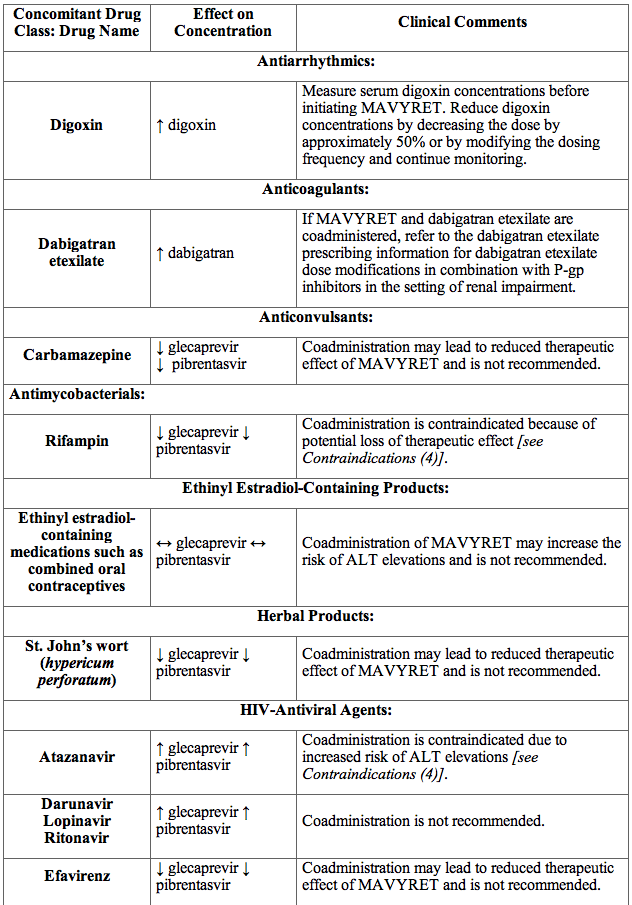
Table 5 provides the effect of MAVYRET on concentrations of coadministered drugs and the effect of coadministered drugs on glecaprevir and pibrentasvir
Table 5. Potentially Significant Drug Interactions Identified in Drug Interaction Studies
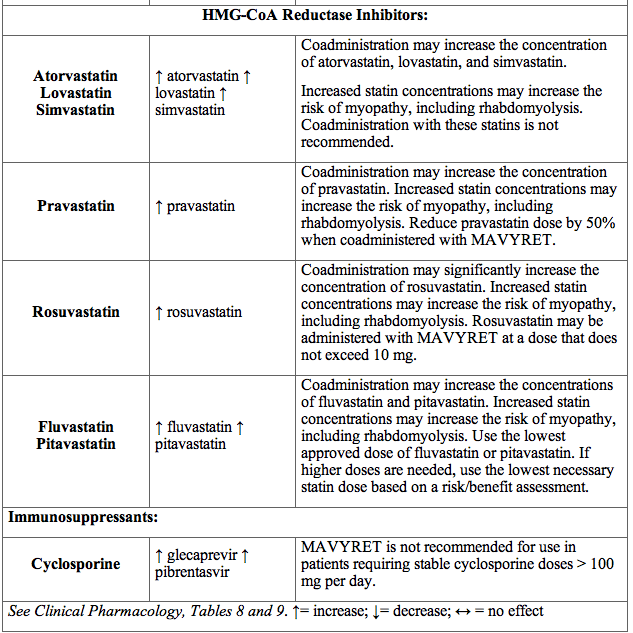
Drugs with No Observed Clinically Significant Interactions with MAVYRET
No dose adjustment is required when MAVYRET is coadministered with the following medications: abacavir, amlodipine, buprenorphine, caffeine, dextromethorphan, dolutegravir, elvitegravir/cobicistat, emtricitabine, felodipine, lamivudine, lamotrigine, losartan, methadone, midazolam, naloxone, norethindrone or other progestin-only contraceptives, omeprazole, raltegravir, rilpivirine, sofosbuvir, tacrolimus, tenofovir alafenamide, tenofovir disoproxil fumarate, tolbutamide, and valsartan.
CLINICAL STUDIES
Description of Clinical Trials
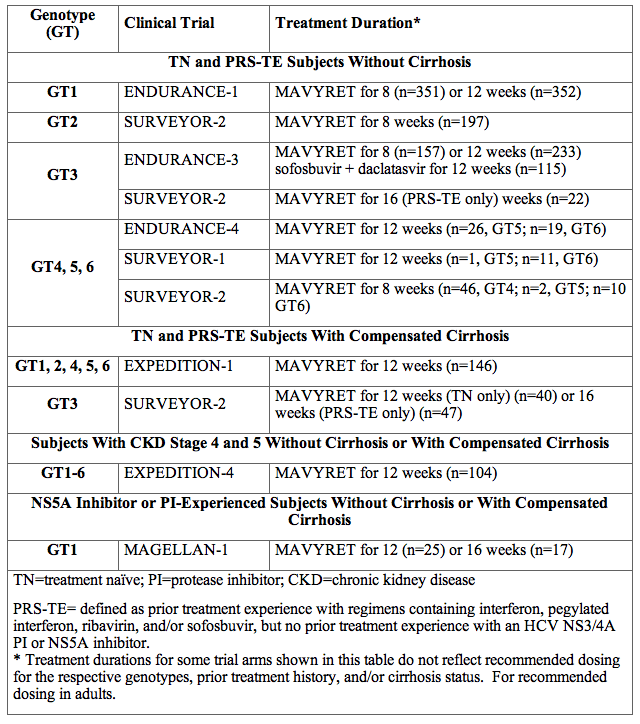
Table 10 summarizes the clinical trials conducted to support the effectiveness of MAVYRET in subjects with HCV genotype 1, 2, 3, 4, 5 or 6 infection and compensated liver disease (including Child-Pugh A cirrhosis) according to treatment history and cirrhosis status.
Table 10. Clinical Trials Conducted with MAVYRET in Adults With HCV Genotype 1, 2, 3, 4, 5 or 6 Infection and Compensated Liver Disease
Serum HCV RNA values were measured during the clinical trials using the Roche COBAS AmpliPrep/COBAS Taqman HCV test (version 2.0) with a lower limit of quantification (LLOQ) of 15 IU/mL (except for SURVEYOR-1 and SURVEYOR-2 which used the Roche COBAS TaqMan real-time reverse transcriptase-PCR (RT-PCR) assay v. 2.0 with an LLOQ of 25 IU/mL). The primary endpoint across all clinical trials was sustained virologic response (SVR12), defined as HCV RNA less than LLOQ at 12 weeks after the end of treatment. Relapse was defined as HCV RNA ≥ LLOQ after end-of-treatment response among subjects who completed treatment. Subjects with missing HCV RNA data, such as those who discontinued due to an adverse event, subject withdrawal or were lost to follow-up, were counted as SVR12 failures.
Demographics and Baseline Characteristics of Clinical Trials in Treatment-Naïve or Treatment-Experienced Adults to PegInterferon, Ribavirin and/or Sofosbuvir (PRS) Without Cirrhosis or With Compensated Cirrhosis (Child-Pugh A)
Of the 2,152 treated subjects without cirrhosis or with compensated cirrhosis who were treatment-naïve or treatment-experienced to a combination of interferon, peginterferon, ribavirin and/or sofosbuvir (PRS), excluding EXPEDITION-4 and MAGELLAN-1, the median age was 54 years (range: 19 to 88); 73% were treatment-naïve, 27% were PRS treatment-experienced; 39% were HCV genotype 1; 21% were HCV genotype 2; 29% were HCV genotype 3; 7% were HCV genotype 4; 4% were HCV genotype 5-6; 13% were ≥65 years; 54% were male; 5% were Black; 12% had cirrhosis; 20% had a body mass index of at least 30 kg per m2; and median baseline HCV RNA level was 6.2 log10 IU/mL.
Treatment-Naïve or PRS Treatment-Experienced Adults With HCV Genotype 1, 2, 4, 5, or 6 Infection Without Cirrhosis
The efficacy of MAVYRET in subjects who were treatment-naïve or treatment-experienced to combinations of peginterferon, ribavirin and/or sofosbuvir (PRS) with genotype 1, 2, 4, 5 or 6 chronic HCV infection without cirrhosis was studied in four trials using 8- or 12-week durations: ENDURANCE-1, ENDURANCE-4, SURVEYOR-1 (Part 2), and SURVEYOR-2 (Part 2 and Part 4).
ENDURANCE-1 was a randomized (1:1), open-label, multi-national trial comparing the efficacy of 8 weeks of treatment with MAVYRET versus 12 weeks of treatment in subjects without cirrhosis with genotype 1 infection with or without HIV-1 co-infection (n=33 co-infected). Table 11 presents SVR12 in MAVYRET-treated genotype 1 infected subjects for the 8 week treatment arm. Due to numerically similar efficacy, MAVYRET is recommended for 8 weeks for treatment-naïve and PRS treatment-experienced genotype 1 subjects without cirrhosis, rather than 12 weeks.
Table 11. ENDURANCE-1: Efficacy in Treatment-Naïve and PRS Treatment-Experienced With HCV Genotype 1 Infection and Without Cirrhosis

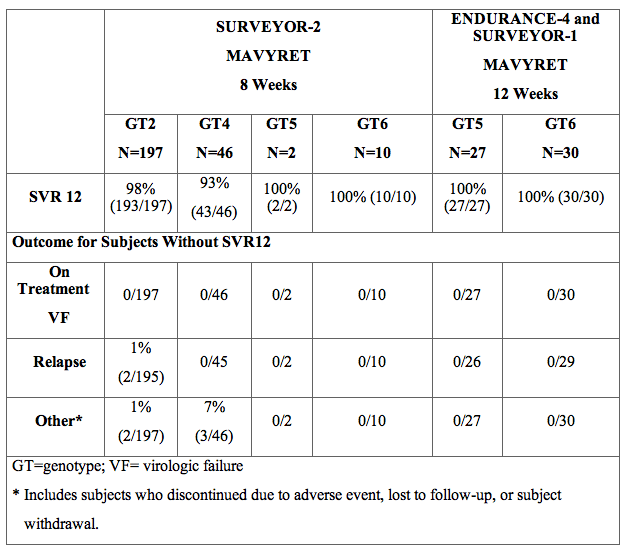
The SVR12 data from the open-label trials SURVEYOR-2 (Parts 2 and 4), ENDURANCE-4 and SURVEYOR-1 (Part 2) are pooled by genotype, where appropriate, in Table 12 for ease of display.
Table 12. SURVEYOR-2 (Part 2 and Part 4), ENDURANCE-4 and SURVEYOR-1 (Part 2): Efficacy in Treatment-Naïve and PRS Treatment-Experienced Adults With HCV Genotypes 2, 4, 5 or 6 Infection Without Cirrhosis
Treatment-Naïve or PRS Treatment-Experienced Adults with HCV Genotype 1, 2, 4, 5, or 6 Infection With Compensated Cirrhosis.
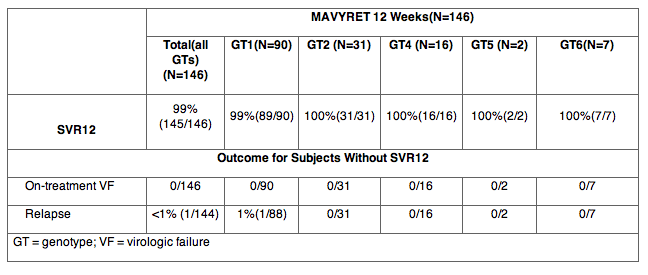
The efficacy of MAVYRET in subjects who were treatment-naïve or treatment-experienced to combinations of peginterferon, ribavirin and/or sofosbuvir (PRS) with genotype 1, 2, 4, 5 or 6 chronic hepatitis C virus infection with compensated cirrhosis (Child-Pugh A) was studied in the single-arm, open-label EXPEDITION-1 trial, which included 146 subjects treated with MAVYRET for 12 weeks.
Table 13. EXPEDITION-1: Efficacy in Treatment-Naïve and PRS Treatment-Experienced Adults With HCV Genotype 1, 2, 4, 5 or 6 Infection With Compensated Cirrhosis
Treatment-Naïve or PRS Treatment-Experienced Adults With HCV Genotype 3 Infection Without Cirrhosis or With Compensated Cirrhosis
The efficacy of MAVYRET in subjects without cirrhosis or with compensated cirrhosis who were treatment-naïve or treatment-experienced to combinations of peginterferon, ribavirin and/or sofosbuvir (PRS) with genotype 3 chronic HCV infection was studied in ENDURANCE-3 and in SURVEYOR-2 Part 3.
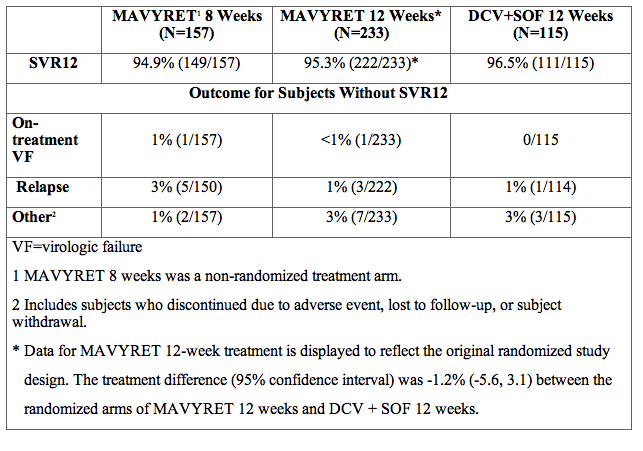
ENDURANCE-3 was a partially-randomized, open-label, active-controlled trial in treatment-naïve subjects. Subjects were randomized (2:1) to either MAVYRET for 12 weeks or to the combination of sofosbuvir and daclatasvir for 12 weeks; subsequently the trial included a third non-randomized arm with MAVYRET for 8 weeks. The SVR12 data are summarized in Table 14. Due to numerically similar efficacy MAVYRET is recommended for 8 weeks for treatment-naïve genotype 3 subjects without cirrhosis, rather than 12 weeks.
Table 14. ENDURANCE-3: Efficacy in Treatment-Naïve, HCV Genotype 3-Infected Subjects Without Cirrhosis
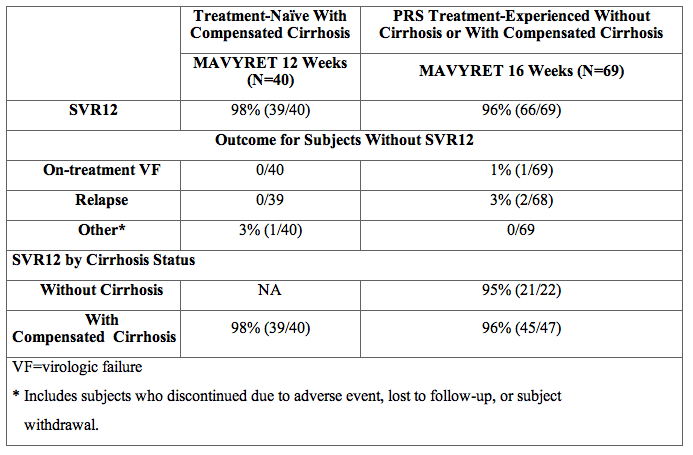
SURVEYOR-2 Part 3 was an open-label trial randomizing PRS treatment-experienced subjects with genotype 3 infection without cirrhosis to 12- or 16-weeks of treatment. In addition, the trial evaluated the efficacy of MAVYRET in genotype 3 infected subjects with compensated cirrhosis in two dedicated treatment arms using 12-week (treatment-naïve only) and 16-week (PRS treatment-experienced only) durations. Among PRS treatment-experienced subjects treated with MAVYRET for 16 weeks, 49% (34/69) had failed a previous regimen containing sofosbuvir.
Table 15. SURVEYOR-2 Part 3: Efficacy in Treatment-Naïve or PRS Treatment-Experienced, HCV Genotype 3-Infected Adults Without Cirrhosis or With Compensated Cirrhosis
Treatment-Naïve and PRS Treatment-Experienced Adults With CKD Stage 4 and 5 and Chronic HCV Infection Without Cirrhosis or With Compensated Cirrhosis
EXPEDITION-4 was an open-label, single-arm, multicenter trial to evaluate safety and efficacy in subjects with severe renal impairment (CKD Stages 4 and 5) with compensated liver disease (with and without Child-Pugh A cirrhosis). There were 104 subjects enrolled, 82% were on hemodialysis, and 53%, 15%, 11%, 19%, 1% and 1% were infected with HCV genotypes 1, 2, 3, 4, 5 and 6; respectively. Overall, 19% of subjects had compensated cirrhosis and 81% of subjects were non-cirrhotic; 58% and 42% of subjects were treatment-naïve and PRS treatment-experienced, respectively. The overall SVR12 rate was 98% and no subjects experienced virologic failure. The presence of renal impairment did not affect efficacy; no dose-adjustments were required during the trial.
Adults Who are NS5A Inhibitor or NS3/4A-Protease Inhibitor (PI)-Experienced, Without Cirrhosis or With Compensated Cirrhosis
MAGELLAN-1 was a randomized, multipart, open-label trial in 141 genotype 1- or 4-infected subjects who failed a previous regimen containing an NS5A inhibitor and/or NS3/4A PI. Part 1 (n=50) was a randomized trial exploring 12 weeks of glecaprevir 200 mg and pibrentasvir 80 mg, glecaprevir 300 mg and pibrentasvir 120 mg, with and without ribavirin (only data from glecaprevir 300 mg plus pibrentasvir 120 mg without ribavirin are included in these analyses). Part 2 (n=91) randomized genotype 1- or 4-infected subjects without cirrhosis or with compensated cirrhosis to 12- or 16-weeks of treatment with MAVYRET.
Of the 42 genotype 1-infected subjects treated in Parts 1 and 2, who were either NS5A inhibitor-experienced only (and treated for 16 weeks), or NS3/4A PI-experienced only (and treated for 12 weeks), the median age was 58 years (range: 34 to 70); 40% of the subjects were NS5A-treatment experienced only and 60% were PI experienced only; 24% had cirrhosis; 19% were ≥65 years, 69% were male; 26% were Black; 43% had a body mass index ≥ 30 kg/m2; 67% had baseline HCV RNA levels of at least 1,000,000 IU per mL; 79% had subtype 1a infection, 17% had subtype 1b infection and 5% had non-1a/1b infection.
Due to higher rates of virologic failure and treatment-emergent drug resistance, the data do not support labeling for treatment of HCV genotype 1 infected patients who are both NS3/4A PI and NS5A inhibitor-experienced.
Table 16. MAGELLAN-1: Efficacy in HCV Genotype 1-Infected Adults Who Are NS3/4A PI-Experienced or NS5A Inhibitor-Experienced, Without Cirrhosis or With Compensated Cirrhosis

The updated labels will soon be available drugs@fda or DailyMed
Steve Morin
Office of Health and Constituent Affairs
Food and Drug Administration
Richard Klein
Office of Health and Constituent Affairs
Food and Drug Administration
Kimberly Struble
Division of Antiviral Products
Food and Drug Administration
For more information about the Hepatitis Liaison Program visit the FDA Patient Network
|
|
| |
| |
|
|
|