| |
Public-Private Partnership: Targeting Real-World Data for Hepatitis C Direct-Acting Antivirals [HCV-TARGET]
|
| |
| |
Gastroenterology Sept 2017
Download the PDF here
Poonam Mishra, Jeffry Florian
US Food and Drug Administration, Silver Spring, Maryland
Joy Peter
University of Florida, Gainesville, Florida
Monika Vainorius, Michael W. Fried
University of North Carolina, Chapel Hill, North Carolina
David R. Nelson
University of Florida, Gainesville, Florida
Debra Birnkrant
US Food and Drug Administration, Silver Spring, Maryland
The era of direct-acting antivirals (DAAs) has transformed the treatment landscape for chronic hepatitis C virus (HCV) infection. Primarily this is due to the approval of multiple DAAs that are highly efficacious with improved safety profiles that became available within a short time span.1 Regulatory agencies have a public health responsibility to ensure the safety and efficacy of approved drug products. Although the demonstrated safety and efficacy of the drugs in registrational trials is paramount, the continued safety and effectiveness of drugs and treatment outcomes in diverse clinical care settings after a drug's approval are of great importance. This paper describes an innovative collaborative platform using real-world clinical practice settings to gather safety and effectiveness data for DAAs approved for the treatment of chronic HCV infection.
Background
The US Food and Drug Administration (FDA) strives to balance the timely access of novel therapies to patients in need with gathering additional data in subgroups in the postmarketing phase. A current approach is to request drug sponsors to conduct postmarketing phase IV studies or clinical trials to expand our knowledge and understanding of novel therapies. However, sometimes results from these postmarketing studies and trials may be outdated by the time final results become available owing to rapid advancements in the scientific field. This was particularly observed with first-generation HCV DAAs, boceprevir and telaprevir, which received regulatory approval in 2011. By the time some of the postmarketing trials for these drugs were completed, the results were outdated because the HCV treatment landscape rapidly moved into the interferon-free era.2 In such a rapidly evolving treatment landscape, innovative solutions are required to obtain "real-world data" at the earliest time points feasible.
An alternative approach can involve use of a systematic observational cohort evaluating new drugs or therapies that leverage real-world evidence. This approach could help the FDA to strike a balance between premarket evaluation and postmarket data collection to facilitate identification of emerging safety signals in the postmarketing setting. Using real-world evidence to enhance the safety and effectiveness of new drugs can be achieved through robust public-private partnerships. This approach has been used by the FDA's Center for Drug Evaluation and Research, Division of Antiviral Products in partnership with Hepatitis C Therapeutic Registry and Research Network (HCV-TARGET).3
There are specific disease characteristics of chronic hepatitis C and its treatment that make it amenable to using real-world evidence for further informing safety and effectiveness. First, chronic hepatitis C is a disease that progresses over a long period of time and, if left untreated, chronic HCV infection is rarely associated with spontaneous cure.4 As such, there is a negligible placebo effect and clearance of the virus is attributed to the treatment intervention. Second, the clinical assessment of virologic cure, sustained virologic response (SVR), is an objective and reliable endpoint of treatment efficacy that correlates with improvement in clinical outcomes and is routinely assessed by practicing physicians.5 Finally, chronic hepatitis C treatment durations are relatively short and highly effective interventions, limiting the extent of missing data from real-world observational studies.
The FDA and HCV-TARGET
HCV-TARGET is a cooperative academic consortium that is partially supported through a National Institutes of Health Clinical and Translational Science Award, augmented with substantial support from pharmaceutical sponsors. The organization, headed by a Steering Committee of hepatology experts including a Clinical Coordinating Center based at the University of Florida and a Data Coordinating Center based at the University of North Carolina, has forged key partnerships between academic centers, community sites, and private industry (Figure 1). The Steering Committee provides oversight and guidance to HCV-TARGET. The HCV-TARGET Industry Advisory Council, composed of 1 representative from each industry sponsor, serves in a nonvoting advisory capacity to the Steering Committee and provides input on the use of network data and new initiatives consistent with the goals of HCV-TARGET. The FDA Advisory Council advises the HCV-TARGET Network as part of their ongoing charge to protect and improve public health by ensuring the safety, and efficacy of drugs, biological products, and medical devices related to HCV. The Community Advocate Representatives provide input on the needs and ideas of the HCV community in framing new initiatives and long-term goals for HCV-TARGET. A formal memorandum of understanding formed the basis for development of scientific collaborations, outreach, and educational initiatives, and intellectual partnerships between FDA and HCV-TARGET. Last, the Publications Committee oversees the activities of the scientific publications and presentations of HCV-TARGET. This committee assures the appropriate public dissemination of HCV-TARGET Network data, the completion of manuscripts, and adherence to principles of authorship and conflicts.
HCV-TARGET established a common research database to be able to conduct a longitudinal observational study to evaluate use of approved DAAs for the treatment of hepatitis C in clinical practice to (1) rapidly inform strategies for better management of populations represented and underrepresented in clinical trials, (2) identify and remediate gaps relative to treatment guidelines, (3) describe adverse event management to optimize treatment, and (4) serve as the core resource for collaborative translational studies using biospecimens and clinical data from diverse patient populations. Through this collaboration, the FDA has access to the HCV-TARGET resources to further inform use of newly approved HCV drugs in an actual clinical practice setting. The collaboration provides a robust platform for FDA scientists to learn from study data to inform areas for improvement in clinical trial design. The HCV-TARGET model allows rapid data acquisition across multiple regimens being used in a disease population receiving care in routine clinical practice. Thus, the study design is disease focused, and not drug specific. This allows for continuous acquisition of data as new drugs enter the market.
Data Collection Methods and Standards
HCV-TARGET uses innovative approaches related to bioinformatics, epidemiology, biostatistics, and health care data systems integration. Patients at participating sites are consented prospectively for participation. HCV-TARGET uses a centralized data abstraction process that minimizes burden on research sites and reduces data variability owing to difference in interpretation of medical records. The site redacts protected health information from the entire electronic medical record and transmits it electronically to the Clinical Coordinating Center where the data are entered in a standardized format into the HCV-TARGET research electronic data capture database. The database was established to be compliant with Title 21of the Code of Federal Regulations Part 11 governing electronic records and adherent to Clinical Data Interchange Standards Consortium standards that facilitate data exchange to both sponsors and the FDA. HCV-TARGET's comprehensive, observational cohort uses meticulous data collection methods that have <2% missing data elements and <5% loss to follow-up.
Notable Outcomes
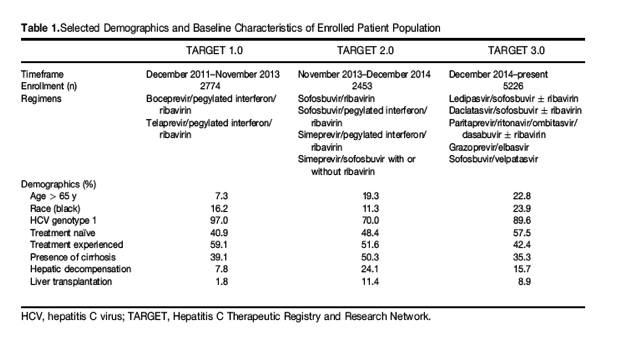
Since inception in 2011, HCV-TARGET has enrolled >10,000 patients treated with HCV DAA-based regimens approved by the FDA (Table 1). The HCV-TARGET protocol allows for enrollment of populations that may have been underrepresented in premarket clinical trials; this data may be useful in understanding the safety and/or effectiveness in these subpopulations. Thus, patients from underrepresented minorities, those with cirrhosis, decompensated cirrhosis, and those undergoing liver transplant, for example, have been oversampled compared with the general population of patients treated for hepatitis C and those enrolled in clinical trials (Table 1).
Boceprevir and Telaprevir
HCV-TARGET analyzed the experiences of >2000 patients who were among the first patients in the United States to receive triple therapy with telaprevir or boceprevir in combination with pegylated interferon (PEG-IFN) and ribavirin.6, 7 The results demonstrated that patients treated in clinical practice with these agents had high rates of advanced disease (38% cirrhosis), had lower SVR rates, and were more likely to sustain significant adverse events than participants in the registrational trials. Rates of anemia and treatment discontinuations in the HCV-TARGET analysis were higher than reported for the pivotal registrational trials, although the HCV-TARGET population had more advanced liver disease.8, 9, 10, 11 The lower SVR rates in HCV-TARGET could be explained by the higher proportion of patients with cirrhosis and of African American patients, factors that have all been associated with lower SVR.12
Sofosbuvir and Simeprevir
The usefulness of the HCV-TARGET model was apparent in 2013 when 2 new "triple therapy" regimens became available: simeprevir in combination with PEG-IFN and ribavirin, and sofosbuvir in combination with PEG-IFN and ribavirin. Given that PEG-IFN, the backbone of HCV treatment for more than a decade, had numerous side effects and contraindications for use in many patients, physicians were seeking all-oral, well-tolerated, and highly effective alternatives. A small phase II study that combined simeprevir plus sofosbuvir, without PEG-IFN, demonstrated remarkably high SVR rates with only 12 to 24 weeks of therapy in difficult-to-cure populations.13 Shortly thereafter, the "off-label" all-oral regimen of simeprevir plus sofosbuvir became one of the most frequently prescribed regimens for patients in the United States. The HCV-TARGET network accrued data on safety and effectiveness of this unapproved regimen in nearly 1000 patients in routine clinical practice.14
The FDA also became aware of the extent of "off-label" use of the simeprevir and sofosbuvir combination regimen and reviewed the FDA's Adverse Event Reporting System database to identify any safety signals for this off-label combination use. However, a major limitation of the Adverse Event Reporting System is a reliance on passive reporting of adverse events by health care professionals or consumers, which unfortunately may include incomplete information on relevant patient characteristics, baseline disease information, comorbid conditions, or concomitant medications.15 Selective reporting also makes it difficult to assess the incidence of unexpected adverse events when medications are used in a broader population outside of clinical trials.
Coinciding with this "off-label" use of the simeprevir and sofosbuvir combination regimen, an efficacy supplement was submitted to the FDA in 2014 to support the use of these 2 drugs together based on the previously mentioned phase II results.16 The sponsor approached the HCV-TARGET group to obtain the overall safety and effectiveness data evaluated in the HCV-TARGET database and submitted the summary data to the FDA to provide supportive evidence and reassurance in terms of safety of the combination regimen. Based on a comprehensive review of the available data, the simeprevir indication was expanded to include its use in combination with sofosbuvir providing an all-oral treatment option for patients. Final results from 2 phase III confirmatory trials, specifically submitted to fulfill the postmarketing commitments, are comparable with the results generated from HCV-TARGET in a real-world setting.17, 18
Sofosbuvir and Ledipasvir
The HCV-TARGET consortium continued to accrue data as other approved therapeutic regimens entered clinical practice. A phase III study of sofosbuvir plus ledipasvir suggested that patients with favorable treatment characteristics at baseline when treated for an 8-week duration had similar SVR rates compared with those treated for 12 weeks.19 FDA labeling of the sofosbuvir plus ledipasvir regimen states that treatment for 8 weeks could be considered for patients who were genotype 1 without cirrhosis, treatment naïve, with pretreatment HCV RNA viral load of <6 million IU/mL. As a cost savings measure, payers frequently mandated the shortened duration regimen, which raised concerns for practitioners, particularly when patients had advanced fibrosis or levels of viremia at the upper boundary of 6 million IU/mL. HCV-TARGET performed a detailed analysis of patients who met the labeled criteria for shortened duration therapy and who subsequently received only 8 weeks treatment compared with similar patients who received 12 weeks of therapy.20 The SVR12 rate was 96% (95% CI, 94-99) in the group that received 8 weeks (244/255) and 98% (95% CI, 95-99) in the group that received 12 weeks (289/296). These results suggest that the shortened duration of treatment in usual clinical practice paralleled the results obtained in phase III clinical trials.
Specific Populations
Often, the safety and efficacy data to inform treatment decisions in specific subpopulations such as transplant recipients or those with advanced liver disease such as decompensated liver disease becomes available at some point after approval. The HCV-TARGET network was instrumental in systematically collecting these data as clinicians were using these regimens after approval in subpopulations with unmet needs (Table 1).21, 22, 23, 24 From the data on early DAA regimens assessed in HCV-TARGET database, treatment for genotype 3 patients with decompensated cirrhosis was identified as an unmet need.22
Monitoring of Serious Adverse Events
The FDA is committed to ensuring safety throughout the life cycle of a drug from premarket testing and clinical development through postmarketing surveillance and risk management.25 The FDA may identify a new safety signal or identify more serious or more frequent reports of a known safety risk from various sources during the postmarketing period once a drug is used in a real-world setting. The HCV-TARGET platform characterizes and tabulates all adverse events abstracted from redacted medical records provided by clinicians throughout treatment and posttreatment follow-up. Serious adverse events are monitored and queried to generate a complete understanding of the event and the results are reported to sponsors and the FDA. Both regulatory authorities and sponsors have access to HCV-TARGET resources that have been used to evaluate reports of unexpected adverse events for a variety of situations, including (1) the interactions of sofosbuvir and amiodarone causing bradyarrhythmia, (2) the safety of specific regimens in patients with decompensated cirrhosis, (3) the prevalence of baseline resistant-associated variants in usual clinical practice, and (4) the incidence of hepatocellular carcinoma recurrence after SVR with a DAA regimen. Thus, HCV-TARGET uses an evidence-based approach to evaluate the postmarketing experience of these drugs. These data are reviewed as part of the comprehensive assessment of the identified safety risk.
In general, the interpretation of postmarketing safety data involves a benefit-risk assessment based on the review of data from multiple sources such as spontaneous adverse event reports, observational studies, clinical trials, published literature, and estimates of drug usage and background rates of the adverse event. The relevant data are interpreted in the context of the known safety and efficacy profiles, as demonstrated in clinical trials supporting approval. The totality of the existing information is taken into consideration when assessing the public health risks of an approved drug. Based on appropriate scientific data, the FDA may require certain postmarketing studies and clinical trials to further assess a known serious risk, and/or to assess signals of serious risk, and/or to identify an unexpected serious risk when available data indicate the potential for a serious risk.26 Nontraditional sources of information, such as that generated by HCV-TARGET, have demonstrated usefulness as an important component in informing the postmarketing safety of new HCV treatments.
Limitations
There are several limitations to HCV-TARGET database. First, there is no control group of untreated hepatitis C patients. Without an unexposed control group, the causality assessment of adverse outcomes cannot always be ascertained; uncertainty may remain as to whether the observed adverse reactions are due to the treatment regimen, progression of underlying disease, other comorbid conditions and/or effects secondary to concomitant medications. Second is the issue of selection bias; it cannot be determined how the proportion of patients who enrolled (received treatment) is different from nonenrollees who did not receive treatment. In addition, the demographics of the enrolled population may change over time based on criteria used by third-party payers for coverage of available treatment options.
Moving Forward
Comparative effectiveness trials are important and informative to compare the safety profile and the efficacy of various approved regimens. Although the approved DAA regimens have demonstrated robust efficacy and reasonable safety across a range of genotypes, most of the regimens have been approved using placebo-controlled, historic-controlled, or dose- and duration-controlled trials with limited direct comparison against other approved regimens. This information gap is important for practitioners who may be interested in a direct comparison between these approved regimens to best inform the ideal regimen based on a patient demographics and viral genotype and subtype. When comparative trials are not feasible to conduct before approval, effectiveness data can be collected postmarketing as demonstrated in HCV-TARGET database.
This use of systematically collected observational cohort data to demonstrate clinical effectiveness holds promise. Real-world effectiveness data could provide supportive evidence if included as a component of an overall benefit-risk assessment with other studies and can contribute to the totality of data available for regulatory decision making. Longitudinal follow-up cohort studies provide a means to assess long-term outcomes in the setting of SVR and obtain much needed safety data once a drug is used more widely and under more diverse conditions in a real-world setting. The data generated will help to provide important safety and effectiveness information on how the drugs performed in the more diverse populations in which they are intended for use. In addition, the results may further guide providers and patients to assure safe and effective use of drugs. This public-private partnership provides a neutral platform for academia, pharmaceutical companies, and regulatory agencies to collaborate and work effectively to reach mutual goals of responding to public health needs in a timely manner.
| |
| |
| |
|
|
|