| |
Glecaprevir plus pibrentasvir for chronic hepatitis C virus genotype 1, 2, 4, 5, or 6 infection in adults with compensated cirrhosis (EXPEDITION-1): a single-arm, open-label, multicentre phase 3 trial
|
| |
| |
Download the PDF here
Lancet Infect Dis August 14, 2017 - Xavier Forns, Samuel S Lee, Joaquin Valdes, Sabela Lens, Reem Ghalib, Humberto Aguilar, Franco Felizarta, Tarek Hassanein, Holger Hinrichsen,
Diego Rincon, Rosa Morillas, Stefan Zeuzem, Yves Horsmans, David R Nelson, Yao Yu, Preethi Krishnan, Chih-Wei Lin, Jens J Kort, Federico J Mensa
Summary
Background
The once-daily, ribavirin-free, pangenotypic, direct-acting antiviral regimen, glecaprevir coformulated with pibrentasvir, has shown high rates of sustained virological response in phase 2 and 3 studies. We aimed to assess the efficacy and safety of 12 weeks of coformulated glecaprevir and pibrentasvir in patients with hepatitis C virus (HCV) infection and compensated cirrhosis.
Methods
We did this single-arm, open-label, multicentre phase 3 study at 40 sites in Belgium, Canada, Germany, South Africa, Spain, and the USA. We enrolled patients aged 18 years or older with HCV genotype 1, 2, 4, 5, or 6 infection and compensated cirrhosis. Patients were either HCV treatment-naive or had not responded to treatment with interferon or pegylated interferon with or without ribavirin, or sofosbuvir plus ribavirin with or without pegylated interferon. Oral glecaprevir (300 mg) coformulated with pibrentasvir (120 mg) was administered once daily for 12 weeks. The primary efficacy endpoint was sustained virological response at post-treatment week 12 (HCV RNA <15 IU/mL). We assessed efficacy and safety in all patients who received at least one dose of study drug (intention-to-treat population). This study is registered with ClinicalTrials.gov, number NCT02642432.
Findings
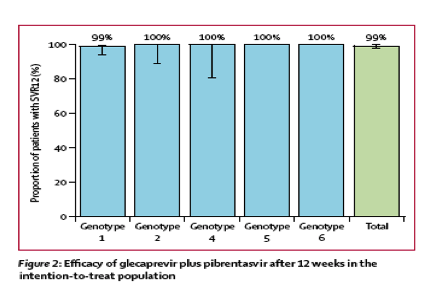
Between Dec 7, 2015, and May 4, 2016, we enrolled 146 patients with compensated cirrhosis, of whom 48 (33%) had genotype 1a HCV infection, 39 (27%) had genotype 1b infection, 34 (23%) had genotype 2 infection, 16 (11%) had genotype 4 infection, two (1%) had genotype 5 infection, and seven (5%) had genotype 6 infection. 12 weeks after treatment, 145 patients (99%, 95% CI 98-100) achieved sustained virological response, with one (1%) relapse at post-treatment week 8. We recorded 101 (69%) adverse events, of which 65 (64%) were mild. The most common adverse events were fatigue (n=28 [19%]) and headache (n=20 [14%]). 11 (8%) patients had serious adverse events, none of which were deemed related to study drugs. No patients had elevations in alanine aminotransferase and no patients prematurely discontinued treatment because of adverse events.
Interpretation
Our results show that 99% of patients treated with once-daily glecaprevir plus pibrentasvir achieved a sustained virological response at 12 weeks. Furthermore, this drug regimen had a favourable safety profile in previously treated or untreated patients with chronic HCV genotype 1, 2, 4, 5, or 6 infection and compensated cirrhosis. These findings could help simplify treatment algorithms and reduce treatment burden.
Funding
AbbVie.
Introduction
Hepatitis C virus (HCV) infection affects 72-185 million individuals worldwide1, 2, 3 and, when left untreated, chronic infection can lead to liver cirrhosis. In the absence of therapy, the number of HCV-infected patients with cirrhosis is projected to rise over the next decade.4 Patients with cirrhosis are at increased risk for development of hepatocellular carcinoma, and those with hepatocellular carcinoma who do not undergo liver transplantation have a 5 year survival rate of 50%.5, 6 The incidence of HCV infection continues to increase worldwide, and every year an estimated 500 000 people die of HCV-related diseases.7 Six major genotypes and 67 subtypes of HCV have been identified.8 Genotype 1 is the most prevalent in the USA (78%) and has an estimated worldwide prevalence of 46%. Genotypes 2, 3, 4, 5, and 6 have global prevalence rates of roughly 9%, 30%, 8%, 0⋅8%, and 5%, respectively.2, 9
Combinations of direct-acting antiviral drugs for targeting multiple components of the HCV viral replication process, including HCV non-structural (NS) proteins 3/4A, 5A, and 5B, have shown high rates of sustained virological response and favourable tolerability.10 However, most guideline-recommended treatment options are not equally effective across all genotypes, and some can require addition of ribavirin or extension of the treatment duration up to 24 weeks to increase efficacy for patients with compensated cirrhosis.11, 12 Because longer treatment durations often require additional on-treatment monitoring, and use of ribavirin is contraindicated for several medical conditions, a regimen that overcomes these requirements could make treatment available to a broader population of patients and physicians. A pangenotypic, direct-acting antiviral HCV treatment for patients with compensated cirrhosis that maintains high efficacy across all major HCV genotypes without the need for baseline genotyping, viral load, or resistance testing is desirable and might further simplify treatment algorithms.13
Research in context
Evidence before this study
We searched PubMed and meeting abstracts from the European Association for the Study of the Liver (EASL) and the American Association for the Study of Liver Diseases (AASLD) from Nov 1, 2015, to April 1, 2017 for late-stage clinical trials done in patients with hepatitis C virus (HCV) genotype 1-6 infection and cirrhosis treated with pegylated interferon-free direct-acting antiviral drugs. We used the search terms “HCV or hepatits C virus” and “cirrho” and “clinical study or trial”. We excluded studies that included pegylated interferon in combination with a direct-acting antiviral. At the time of study design, a pangenotypic treatment for patients with cirrhosis was unavailable.
Direct-acting antiviral treatment options approved for patients with HCV infection and cirrhosis include 12 weeks of sofosbuvir plus velpatasvir for HCV genotypes 1-6, and 12-16 weeks of grazoprevir plus elbasvir for patients with genotype 1 or 4 infection. Sofosbuvir plus ledipasvir, sofosbuvir plus simeprevir, and ombitasvir, paritaprevir, and ritonavir plus dasabuvir, and are only approved in patients with HCV genotype 1 infection. The SURVEYOR-I and SURVEYOR-II phase 2 studies in patients with HCV genotype 1 or 3 infection and compensated cirrhosis showed that treatment with glecaprevir and pibrentasvir for 12 weeks without ribavirin resulted in a sustained virological response rate of 96% and a favourable safety profile.
Added value of this study
Our findings show that treatment with co-formulated glecaprevir plus pibrentasvir once daily for 12 weeks achieved a sustained virological response rate of 99% in 145 of 146 patients with chronic HCV genotype 1, 2, 4, 5, or 6 infection and compensated cirrhosis (patients with genotype 3 infection and cirrhosis were enrolled in a separate study). Notably, there were no elevations in alanine aminotransferase and no treatment discontinuations due to adverse events, which is indicative of the regimen's favourable safety and tolerability profile.
Implications of all the available evidence
The incidence of HCV infection continues to increase worldwide, and estimates place the number of HCV-related deaths at 500 000 per year. When left untreated, chronic HCV infection can lead to liver cirrhosis; patients with cirrhosis are at increased risk for development of hepatocellular carcinoma, and those with hepatocellular carcinoma who do not undergo liver transplantation have a 5 year survival rate of 50%. An interferon-free and ribavirin-free treatment for most patients with HCV infection and compensated cirrhosis might help simplify treatment algorithms and reduce treatment burden.
Glecaprevir (formerly ABT-493)—an NS3/4A protease inhibitor coformulated with pibrentasvir (formerly ABT-530), an NS5A inhibitor—is currently being evaluated as a pangenotypic regimen and has shown a high barrier to resistance and potency against common NS3 and NS5A polymorphisms.14, 15 Phase 2 studies in patients with HCV genotype 1 or 3 infection and compensated cirrhosis treated with glecaprevir and pibrentasvir for 12 weeks without ribavirin resulted in a sustained virological response rate of 96% at 12 weeks after treatment and a favourable safety profile.16
We did the EXPEDITION-1 study to assess the efficacy and safety of 12 weeks of coformulated glecaprevir plus pibrentasvir in adults with chronic HCV genotype 1, 2, 4, 5 or 6 infection and compensated cirrhosis. Patients with compensated cirrhosis and genotype 3 HCV infection were enrolled in a separate study.17
Methods
Study design and participants
We did this single-arm, open-label, phase 3 study at 40 study sites in Belgium, Canada, Germany, South Africa, Spain, and the USA. We enrolled patients aged 18 years or older (no upper limit) if they had chronic HCV infection, defined as a positive anti-HCV antibody status or HCV RNA at least 6 months before screening, a liver biopsy consistent with chronic HCV infection or abnormal alanine aminotransferase (ALT) for at least 6 months before screening. A plasma HCV RNA concentration of 1000 IU/mL or greater was required at the time of screening. Presence of compensated cirrhosis was documented by liver biopsy with a METAVIR (or equivalent) fibrosis score of 4 or Ishak fibrosis score of more than 4; a FibroTest score of 0⋅75 or more with an aminotransferase-to-platelet ratio index of more than 2, or a FibroScan score of 14⋅6 kPa or more; and a Child-Pugh score of 6 or less at screening and no current or past evidence of Child-Pugh B or C classification or clinical history of liver decompensation. Patients were either HCV treatment-naive or had not responded to treatment with interferon or pegylated interferon with or without ribavirin, or sofosbuvir plus ribavirin with or without pegylated interferon therapy; all patients were NS3/4A and NS5A inhibitor-naive. Absence of hepatocellular carcinoma was confirmed by a negative ultrasound, CT scan, or MRI within 3 months before screening or at screening, and a serum alpha-fetoprotein concentration of less than 100 ng/mL at screening. Patients were additionally required to have a body-mass index of 18 kg/m2 or more (no upper limit).
We excluded patients with a positive test result for hepatitis B surface antigen or anti-HIV antibody, or with a cause of liver disease other than chronic HCV infection. Laboratory exclusion criteria were serum albumin of less than 2⋅8 g/dL, ALT and aspartate aminotransferase (AST) of more than ten times the upper limit of normal, calculated creatinine clearance of less than 50 mL/min, haemoglobin of less than 12 g/dL (men) or 11 g/dL (women), a platelet count of less than 60 000 cells per μL, total bilirubin of more than 3⋅0 mg/dL, and an international normalised ratio of more than 2⋅3.
All patients provided written informed consent. The study was done in accordance with the Declaration of Helsinki, and institutional review board approval was obtained for each study site.
Procedures
Oral glecaprevir (300 mg; AbbVie and Enanta) coformulated with pibrentasvir (120 mg; AbbVie; Cork, Ireland) was administered once daily for 12 weeks. We assessed safety and tolerability during the treatment period and 24 weeks after treatment completion for all patients receiving at least one dose of study drug. The appendix shows the study design, including treatment and follow-up periods.
Virological response was assessed by use of serum HCV RNA concentration with the COBAS AmpliPrep/COBAS TaqMan HCV Quantitative Test, version 2.0 (Roche Molecular Systems, Branchburg, NJ, USA), with a lower limit of quantitation of 15 IU/mL. Samples were collected at the screening visit, the day 1 visit, treatment weeks 1, 2, 4, 8, and 12 (or after discontinuation), and post-treatment weeks 2, 4, 8, 12, and 24. We did next-generation sequencing of samples collected from all patients at baseline, and assessed presence of HCV baseline polymorphisms in NS3 and NS5A relative to subtype-specific reference sequence with a 15% detection threshold. Treatment-emergent substitutions were analysed for patients who had virological failure and were defined as substitutions that were not present at baseline but detected at the time of failure at 2% detection threshold, or enriched at the time of failure relative to baseline by at least 20% within the patient's viral population.
Safety was assessed by adverse event monitoring and laboratory tests, which were conducted during the study until 24 weeks after treatment. All adverse events were graded according to Common Terminology Criteria for Adverse Events, version 4⋅0.
Outcomes
The primary efficacy endpoint was sustained virological response at post-treatment week 12 (HCV RNA <15 IU/mL). Secondary endpoints were the proportion of patients with on-treatment virological failure (defined as confirmed increase of >1 log10 IU/mL above nadir during treatment, confirmed HCV RNA ≥100 IU/mL after HCV RNA < lower limit of quantification [LLOQ] during treatment, or HCV RNA ≥LLOQ at the end of treatment with at least 6 weeks of treatment), and the proportion with post-treatment virological relapse (defined as confirmed HCV RNA ≥LLOQ between end of treatment and 12 weeks after the last dose of study drug in patients who completed treatment as planned and who had HCV RNA < LLOQ at the end of treatment).
Statistical analysis
The target number of patients in this study was determined to be approximately 20% of the total number of patients enrolled in other phase 3 trials evaluating coformulated glecaprevir plus pibrentasvir, which is consistent with other HCV studies in which patients with or without cirrhosis are included.18, 19, 20
The primary analysis was done after all enrolled patients completed the post-treatment week 12 visit. Efficacy, safety, and demographic analyses were done in the intention-to-treat population, defined as all patients who received at least one dose of study drug. The proportion of patients meeting the primary and secondary endpoints of the study was established for each endpoint, and a two-sided 95% CI was calculated with the normal approximation to the binomial distribution for the primary endpoint and the Wilson score method for secondary endpoints. Descriptive statistics were provided for categorical variables and mean and SD were provided for continuous variables. All analyses were done in SAS (version 9.3).
This study is registered with ClinicalTrials.gov, number NCT02642432.
Role of the funding source
The funder of the study had a role in study design, data collection, data analysis, and data interpretation; approved the content; and provided medical writing assistance. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Between Dec 7, 2015, and May 4, 2016, we enrolled 146 patients with compensated cirrhosis (figure 1), of whom 48 (33%) patients had genotype 1a HCV infection, 39 (27%) had genotype 1b infection, 34 (23%) had genotype 2 infection, 16 (11%) had genotype 4 infection, two (1%) had genotype 5 infection, and seven (5%) had genotype 6 infection (table 1). Most patients were white, male, and treatment-naive (table 1). Of the 36 treatment-experienced patients, 25 (69%) had failure of previous treatment with interferon or pegylated interferon with or without ribavirin, whereas 11 (31%) had failure of previous treatment with sofosbuvir plus ribavirin with (n=2) or without (n=9) pegylated interferon (table 1). Median HCV RNA at baseline was 6⋅1 log10 IU/mL (range 3⋅1-7⋅4). Of 133 patients with available sequence data, two (2%) had baseline polymorphisms in NS3 only (positions 155, 156, or 168) and 53 (40%) had polymorphisms in NS5A only (positions 24, 28, 30, 31, 58, 92, or 93); two (2%) patients had polymorphisms in both NS3 and NS5A (table 1). The appendix (p 9)) shows the prevalence of each baseline polymorphism in each target. 31 (21%) patients used concomitant proton-pump inhibitors.
In the intention-to-treat population, 145 patients (99%, 95% CI 98-100) achieved sustained virological response at 12 weeks (figure 2). One (1%) patient with HCV genotype 1a infection with a history of non-response to pegylated interferon plus ribavirin relapsed at post-treatment week 8. The patient had no baseline polymorphisms or treatment-emergent substitutions in NS3; in NS5A, Q30R-H58D was present at the time of failure and Y93N was present at baseline and at the time of failure.
101 (69%) patients had adverse events, 64% of which were mild, with fatigue and headache most frequently reported (table 2). Serious adverse events occurred in 11 (8%) patients (table 2, appendix); none were deemed related to study drugs by the investigator. One (1%) patient with oesophageal varices at screening had oesophageal variceal bleeding on day 22 (appendix), with no worsening of hepatic function (no relevant changes in international normalised ratio, albumin or bilirubin; no other clinical events such as encephalopathy or ascites). One (1%) patient with a history of haemophilia died 61 days after completing the last dose of study drug; the cause of death was cerebral haemorrhage deemed by the investigator as unrelated to study drug. No patient discontinued study drug because of adverse events. Two (1%) new cases of hepatocellular carcinoma occurred (appendix), one 8 days after the last dose of study drug and one on treatment day 40.
The patient with an episode of bleeding from oesophageal varices had a concurrent grade 3 decrease in haemoglobin. No other grade 3 or higher decreases in haemaglobin occurred. We recorded no cases of grade 3 or grade 4 ALT (post-nadir), AST, or total bilirubin elevations to levels higher than those at baseline (table 3). Drug exposures for patients with grade 3 laboratory abnormalities were within the range of other patients in this study (data not shown).
Discussion
12 weeks of treatment with once-daily glecaprevir coformulated with pibrentasvir yielded a sustained virological response rate of 99% in patients with HCV genotype 1, 2, 4, 5, or 6 infection and compensated cirrhosis, with no clinically significant safety events or laboratory abnormalities. These results confirm those from phase 2 studies, in which treatment with glecaprevir and pibrentasvir yielded high rates of sustained virological response and had a favourable safety profile in patients with compensated cirrhosis.16 The high 12 week response rate observed in the overall population was achieved regardless of baseline HCV RNA values, treatment history, presence of baseline NS3 or NS5A polymorphisms, or any other baseline factor.
Currently, the only 12 week, pangenotypic HCV treatment option approved for patients with compensated cirrhosis is sofosbuvir plus velpatasvir, which achieved sustained virological response rates of 99% in patients with HCV genotype 1, 2, 4, 5, or 6 infection;21, 22 however, the regimen is not recommended for patients with severe renal impairment and end-stage renal disease. In EXPEDITION-1, 12 weeks of coformulated glecaprevir plus pibrentasvir achieved equally high rates of sustained virological response and, in another phase 3 study assessing glecaprevir plus pibrentasvir,23 there were no virological failures in 20 patients with compensated cirrhosis and chronic stage 4 or 5 kidney disease.
To date, many studies have largely focused on HCV genotype 1 infection, which accounts for less than 50% of HCV infections worldwide. Post-hoc integrated analyses showed that 95% of patients with cirrhosis and genotype 1 infection achieved sustained virological response after 12 weeks of sofosbuvir and ledipasvir,24 and 12 weeks of elbasvir plus grazoprevir yielded sustained virological response rates of 89% and 98% in patients with cirrhosis infected with genotype 1 (94%), 4 (6%), or 6 (<1%) who were treatment-experienced and treatment-naive, respectively.25 In our study, 38% of patients were infected with genotypes other than genotype 1, and all achieved a sustained virological response 12 weeks after treatment.
Other available treatments for this population might require the use of ribavirin or treatment durations of up to 24 weeks. Because ribavirin is teratogenic and can cause haemolytic anaemia, a direct-acting antiviral regimen that does not require ribavirin could improve tolerability and result in fewer drug interruptions and discontinuations. Some direct-acting antiviral regimens that contain NS5A inhibitors, such as sofosbuvir plus ledipasvir or sofosbuvir plus velpatasvir, require restrictions on the co-administration of proton-pump inhibitors because of decreases in plasma concentrations of direct-acting antiviral drugs, which could compromise efficacy.18, 26 In the present study, a post-hoc subgroup analysis showed that of the patients who used concomitant proton-pump inhibitors, all but one (97%) achieved sustained virological response at 12 weeks, suggesting that use of concomitant proton-pump inhibitors had no effect on efficacy (data not shown).
Once-daily glecaprevir coformulated with pibrentasvir was well tolerated by patients with compensated cirrhosis, with most of the adverse events being mild in severity and none resulting in discontinuation of study drug. Few patients had serious adverse events, and no association was shown between rates of serious adverse events and exposure to glecaprevir plus pibrentasvir. No patients had grade 3 or higher elevations in ALT or bilirubin, consistent with findings from other phase 2 and phase 3 studies assessing this regimen, in which grade 3 laboratory abnormalities were scarce.27, 28, 29, 30 The safety of this regimen in other patient subgroups, including patients with decompensated cirrhosis and patients with cirrhosis and co-infection with HIV-1 or hepatitis B virus, remains to be determined because these patients were excluded from this study. However, in other phase 3 studies,30, 31 patients with HCV and HIV-1 co-infection without cirrhosis who were treated with glecaprevir plus pibrentasvir achieved similarly high sustained virological response rates and had a similarly favourable safety profile as those with HCV monoinfection.
One limitation of this study is the exclusion of some patient subgroups, particularly those with genotype 3 infection. In view of the potential need for longer treatment durations, especially for patients with cirrhosis and HCV treatment experience, patients with genotype 3 infection and compensated cirrhosis, including those with previous treatment experience, were enrolled in the SURVEYOR-II, Part 3 study.17 12 weeks of glecaprevir plus pibrentasvir yielded a sustained virological response rate of 98% (n=39/40), with no virological failures in treatment-naive patients with cirrhosis; 16 weeks of glecaprevir plus pibrentasvir yielded a response rate of 96% (n=45/47) in treatment-experienced patients with genotype 3 infection and cirrhosis.17 We also excluded patients with decompensated cirrhosis; a separate study assessing the regimen in this population is needed because these patients have different efficacy and safety profiles that often require longer treatment durations or addition of ribavirin. A second limitation is the relatively low number of patients in some subgroups, particularly in patients with HCV genotype 5 and 6 infection. Although all patients with genotype 5 and 6 infection achieved sustained virological response at 12 weeks, it is difficult to draw conclusions with high statistical accuracy. Patients with these two genotypes are generally difficult to recruit because of their low prevalence rates.2, 9 A third limitation is the single-arm study design. An active-controlled study design was not feasible given the absence of a pangenotypic regimen for patients with compensated cirrhosis at the time of study design, and a placebo-controlled design in this population would have delayed treatment in patients who were in more urgent need of treatment, and was therefore problematic.
Overall, our results show that all-oral, once-daily glecaprevir coformulated with pibrentasvir is an efficacious treatment option for most patients with HCV infection and compensated cirrhosis.
| |
| |
| |
|
|
|