| |
Commonalities Between Vasculature and Bone
|
| |
| |
Download the PDF here
An Osseocentric View of Arteriosclerosis
Circulation Jan 23 2017
Dwight A. Towler, MD, PhD
UT Southwestern Medical Center,
Internal Medicine, Endocrine
"All osteotropic hormones and drugs have vascular actions, and alterations in conduit vessel functions impact skeletal physiology.2Therefore, interventions and investigations targeting either skeletal frailty or arteriosclerotic diseases (including atherosclerosis) must incorporate experimental design that simultaneously assesses conduit vessel physiology (viz, stiffness, fibrosis, calcification), in addition to coronary artery and metabolic bone disease. Without systematically embracing such experimental design, we miss opportunities to improve the cardiovascular health of our increasingly aging and dysmetabolic population."
Introduction
A good number of us have forgotten Jean Georges Chretien Frederic Martin Lobstein, but the observations of this 19th-century surgeon-pathologist are codified in disease nomenclature used today. As an amateur archeologist, he had a particular interest in mineralized tissues, including bones, and Lobstein's detailed autopsies led him to coin the word "osteoporosis" to denote the skeletal frailty afflicting older individuals. In many of these aged subjects, Lobstein also noted a vascular disease process that, in 1829, he called "arteriosclerosis": the hardening of normally compliant conduit arteries.1 As such, Lobstein was among the first to embrace an osseocentric view of arterial disease.
Virchow1 expanded on this idea in 1860, recounted in his lecture series, Cellular Pathology. He highlighted the inflammatory nature of lipid-laden atherosclerotic lesions and noted that beyond calcification, 1 outcome of arteriosclerotic disease was true focal vascular ossification. With recognition that oxidized low-density lipoprotein cholesterol is proinflammatory and pathogenic in vascular disease leading to acute ischemia-and the advent of successful cholesterol-lowering therapies-atherosclerosis looms large in the awareness of modern medicine as 1 type of arteriosclerosis.
However, the broader concepts and consequences of Lobstein's arteriosclerosis-be it from atherosclerosis or fibrosis and medial thickening of conduit arteries with or without calcific sclerosis-are frequently dismissed. This has led to terminology and experimental design that treat atherosclerosis and arteriosclerosis as interchangeable without consideration of the impact of chronic vascular stiffening. Arteriosclerosis impairs Windkessel physiology-the rubbery elasticity of conduit vessels that ensures smooth distal tissue perfusion throughout the cardiac cycle2-and is a powerful contributor to risk for stroke, cognitive impairment, kidney disease, heart failure, and cardiovascular mortality. It is important to note that matrix and mineralization programs can drive arterial stiffness even in the absence of atheroma.2 Therein lies the broader value of the osseocentric view of arterial disease introduced by Lobstein-and the potential to develop novel approaches to mitigate the consequences of arteriosclerosis.
What can we learn from bone biology to inform our understanding of arteriosclerotic disease? First, it highlights some of the key players-cell types, transcription factors, morphogens, hormones, enzymes, and matrix components-that regulate arterial remodeling and vascular stiffening (Figure). During skeletal development, bone is deposited by two primary mechanisms known as endochondral and membranous ossification. The former mediates long bone formation and fracture repair, whereas the latter mineralizes the skull. The same osteogenic processes and cell lineages (mesenchymal, endothelial, and monocyte) control arteriosclerotic mineralization.2 Diabetes mellitus, dyslipidemia, uremia, and advanced age upregulate vascular expression of powerful bone morphogens of the BMP and Wnt gene families. Innate immune responses activated by metabolic and mechanical cues elicit arterial elaboration of these morphogens which, in a paracrine fashion, coordinate osteogenic transcription and matrix mineralization programs that drive arteriosclerosis.2 Linda Demer noted that entraining osteogenic programs to innate immune activity likely conveyed survival advantage with fungal infections by rigidly walling off these tenacious pathogens. The oxylipids and advanced glucosylation products of diabetes mellitus, dyslipidemia, and aging ectopically trigger these osteogenic, pathogen-associated molecular pattern responses in arteries with untoward consequences. Vascular smooth muscle deletion of genes encoding osteoblast transcription factors mitigates arteriosclerotic stiffness and calcification in preclinical disease models. Osteoclast-like cells participate in diseased arterial matrix remodeling, further driving home the similarities.
Osteogenic contributions to arteriosclerotic disease. The fundamental mechanisms controlling matrix remodeling in bone and vasculature are similar and become even more so with matrix mineralization, as this select subset of commonalities reveals. A cell triad encompassing mesenchymal (osteoblast vs smooth muscle), monocytic (osteoclast vs macrophage), and endothelial (bone sinusoid canopy cell vs vascular endothelium) lineages controls orthotopic and ectopic mineral metabolism. However, studies of arteriosclerotic disease highlight that, although mechanisms of bone and vascular mineralization are similar, regulation varies as a function of the specific histoanatomic venue. This truth was first discovered in the skeleton but also holds for arteriosclerotic disease. Certain endocrine cues reciprocally regulate calcified matrix accrual in highly specific skeletal and vascular venues, in part reflecting the reciprocal impact of inflammation on bone (decreased) versus vascular (increased) mineralization. As such, all osteotropic agents have vasculotropic actions, and this relationship must be addressed in any therapy targeting either bone or cardiovascular health.
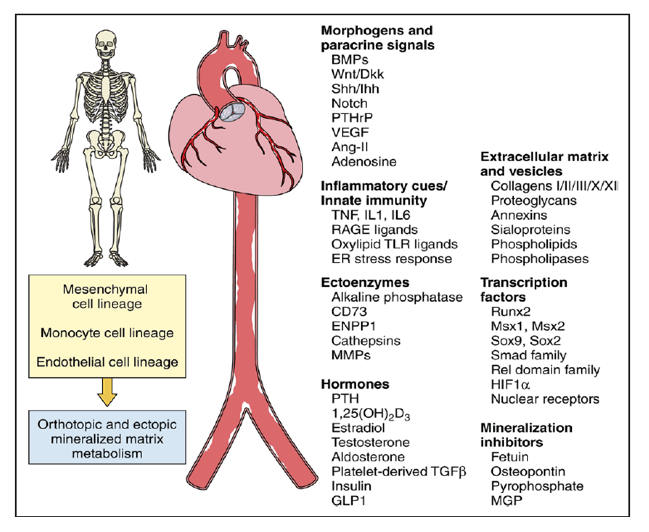
Second, the osseocentric view teaches us that the molecular regulation of matrix calcification differs somewhat depending on the histoanatomic venue. Human genetic studies powerfully extend this lesson to the vasculature.3 St. Hilaire and colleagues3 identified 3 families with profound and precocious lower extremity arteriosclerotic calcification that respected the aortoiliac boundary. Aortic, aortic valve, coronary, carotid, and upper extremity arterial calcium deposition were not observed. Homozygous or compound heterozygous loss-of-function mutations in the ectoenzyme NT5E/CD73 were identified as causal, and the disorder was named arterial calcification of CD73 deficiency The anatomically restricted arteriosclerotic disease of arterial calcification of CD73 deficiency arises in the absence of "traditional" risk factors. Arterial calcification of CD73 deficiency highlights that although commonalities exist, the specifics of arterial matrix calcification and its regulation will differ depending on anatomy and ontogeny, as first taught by bone biologists. Histopathology and pharmacology reinforce these notions in the setting of calcific aortic valve disease. Ectopic endochondral bone replete with hematopoietic elements can be found adjacent to larger concretions of amorphous calcium phosphate, and calcific aortic valve disease is refractory to lipid-lowering strategies that successfully treat coronary artery disease, including calcified plaque regression. Therefore, while sharing some common risk factors, mechanisms and therapeutics identified to inhibit coronary artery calcification and atheroma formation cannot be assumed to favorably impact aortic, aortic valve, and peripheral artery calcification as relevant to the treatment of arteriosclerotic vascular stiffening and its consequences.
Given the similarities in bone formation and vascular calcification, the osseocentric view prompts the following question: how can one hope to apply lessons learned to treatment of arteriosclerosis without exacerbating osteoporosis? Important clues come from the reciprocal impact that certain osteotropic agents have on bone and vasculature mineralization. In preclinical models, intermittent parathyroid hormone receptor signaling can increase bone mass while limiting vascular sclerosis and stiffness.2 In children afflicted with progeria, a precocious aging phenotype characterized by osteoporosis and arteriosclerosis, treatment with the farnesyltransferase inhibitor lonafarnib alone improves bone mass while reducing arterial stiffness-and treatment increases longevity.4 In postmenopausal women, aminobisphosphonates-inhibitors of osteoclast function-improve vertebral bone mass and apparently decrease arterial stiffness and aortic calcification.2 However, drugs such as aminobisphosphonates may negatively impact arterial structure and function in other age groups and sexes (viz, in premenopausal or younger individuals).4 Indeed, even daily dietary calcium supplementation for bone health may exert a sex-specific influence on cardiovascular disease risk.5
Thus, although the osseocentric view lends new therapeutic hope in macrovascular disease management, it clearly highlights the many lessons yet to be learned concerning relationships among vascular biology, arteriosclerosis, and matrix mineralization. All osteotropic hormones and drugs have vascular actions, and alterations in conduit vessel functions impact skeletal physiology.2Therefore, interventions and investigations targeting either skeletal frailty or arteriosclerotic diseases (including atherosclerosis) must incorporate experimental design that simultaneously assesses conduit vessel physiology (viz, stiffness, fibrosis, calcification), in addition to coronary artery and metabolic bone disease. Without systematically embracing such experimental design, we miss opportunities to improve the cardiovascular health of our increasingly aging and dysmetabolic population.
|
|
| |
| |
|
|
|