| |
HIV alters neuronal mitochondrial fission/fusion in the brain during HIV-associated neurocognitive disorders
| | | | |
Download the PDF here
Mitochondrial morphology was preserved in neurons from HIV + brains, with well-formed cristae and undisrupted mitochondria membranes (Fig. 3C). Conversely, mitochondria in HAND neurons were filled with discontinuous cristae, electron-dense structures and damaged double-membranes (Fig. 3D). Analysis of mitochondrial diameter revealed an increase of 80% (p < 0.05) in mitochondrial diameter in HAND brains compared to those from Non-HAND donors. These data suggest that decreased DNM1L in HAND leads to decreased mitochondrial fission and increased fusion.
Overall, these data, along with the accompanying report, reveal a distinct alteration in mitochondrial morphology associated with HIV infection of the CNS and evidence that gp120 mediates these changes via altered mitochondrial fission/fusion and translocation along microtubules. The increase in mitochondria size is associated with severe HAND, and the alterations in fission/fusion machinery are observed early in neuronal models for HIV protein-mediated neurodegeneration. These data suggest that an imbalance in mitochondrial fission/fusion dynamics may be a compensatory mechanism that contributes to neurodegeneration in HAND patients. Furthermore, balancing DNM1L and MFN1 expression levels or activation status may rectify complications observed in GFAP-gp120 tg and other HAND mouse models with the end goal of treating HAND. Future studies will focus on the pathway through which gp120 induces alterations in mitochondrial fission/fusion dynamics.
Analysis of mitochondrial ultrastructure by TEM in gp120 tg and Non-tg animals was performed to corroborate mitochondrial damage observed in our human donor cohort. Neurons in gp120 tg mouse brains contained large and damaged mitochondria with electron dense regions and malformed cristae compared to Non-tg images (Fig. 5C). Diameter measurements revealed that mitochondrial size was significantly increased by 70% in gp120 tg mice (Fig. 5D). These results show that neuronal mitochondria are damaged and elongated in gp120 tg mouse brains.
In the current study we report, for the first time, DNM1L and p-DNM1L-Ser616 protein levels are significantly reduced in brains of HAND donors compared to Non-HAND donors. These changes were reflected in degenerating neurons of HAND donors, and were associated with large and damaged mitochondria. Furthermore, GFAP-gp120 tg mouse brains had decreased DNM1L, increased MFN1, and elongated mitochondria with damaged cristae compared to Non-tg animals. In vitro, gp120 treatment reduced total and active DNM1L expression, increased MFN1, and reduced staining with potentiometric mitochondrial dye (Mitotracker Red) in neuronal cells. Gp120 treatment similarly reduced DNM1L levels in primary rat neurons, concomitant with increased capacity for extracellular acidification. Remarkably, DNM1L overexpression reversed neuroinflammation and neurodegeneration in the GFAP-gp120 tg mice and restored the intensity of Mitotracker staining to levels similar to control in primary rat neurons. Our results indicate that gp120 induced mitochondrial fusion, possibly, as a compensatory, but deleterious response that further exacerbates HIV-induced neuroinflammation and neurodegeneration. These findings are consistent with a recent study that showed DNM1L overexpression reverses HIV protein VPR-induced mitochondrial destruction (Huang et al., 2012).
--------------------
HIV alters neuronal mitochondrial fission/fusion in the brain during HIV-associated neurocognitive disorders
Neurobiology of Disease - Jerel Adam Fields a, Elisabeth Serger a, Sofia Campos b, Ajit S. Divakaruni e, Changyoun Kimb, Kendall Smith a,
Margarita Trejo b, Anthony Adame b, Brian Spencer b, Edward Rockenstein b, Anne N.Murphy e, Ronald J. Ellis b,
Scott Letendre c, Igor Grant d, Eliezer Masliah a,b,⁎
a Department of Pathology, University of California San Diego, La Jolla, CA, USA
b Department of Neurosciences, University of California San Diego, La Jolla, CA, USA
c Department of Medicine, University of California San Diego, La Jolla, CA, USA
d Department of Psychiatry, University of California San Diego, La Jolla, CA, USA
e Department of Pharmacology, University of California San Diego, La Jolla, CA, USA
Highlights
⋅ Reduced mitochondrial fission and increased fusion proteins in brains of HAND donors
⋅ Increased mitochondrial size and damage in brains of HAND donors
⋅ HIV gp120 induces mitochondrial fusion and decreases fission proteins in vivo and in vitro
⋅ Overexpressing mitochondrial fission proteins ameliorates gp120-induced
neurodegeneration in vivo
Abstract
HIV-associated neurocognitive disorders (HAND) still occur in approximately 50% of HIV patients, and therapies to combat HAND progression are urgently needed. HIV proteins are released from infected cells and cause neuronal damage, possibly through mitochondrial abnormalities. Altered mitochondrial fission and fusion is implicated in several neurodegenerative disorders. Here, we hypothesized that mitochondrial fission/fusion may be dysregulated in neurons during HAND. We have identified decreased mitochondrial fission protein (dynamin 1-like; DNM1L) in frontal cortex tissues of HAND donors, along with enlarged and elongated mitochondria localized to the soma of damaged neurons. Similar pathology was observed in the brains of GFAP-gp120 tg mice. In vitro, recombinant gp120 decreased total and active DNM1L levels, reduced the level of Mitotracker staining, and increased extracellular acidification rate (ECAR) in primary neurons. DNM1L knockdown enhanced the effects of gp120 as measured by reduced Mitotracker signal in the treated cells. Interestingly, overexpression of DNM1L increased the level of Mitotracker staining in primary rat neurons and reduced neuroinflammation and neurodegeneration in the GFAP-gp120-tg mice. These data suggest that mitochondrial biogenesis dynamics are shifted towards mitochondrial fusion in brains of HAND patients and this may be due to gp120-induced reduction in DNM1L activity. Promoting mitochondrial fission during HIV infection of the CNS may restore mitochondrial biogenesis and prevent neurodegeneration.
Abbreviations
HAND HIV-associated neurocognitive disorders
DNM1L dynamin 1-like
MFN mitofusin
ECAR extracellular acidification rate
ANI asymptomatic neurocognitive impairment
MND minor neurocognitive disorder
HAD HIV-associated dementia
1. Introduction
The number of human immunodeficiency virus (HIV) cases has increased to over 34 million individuals worldwide (Brun-Vezinet and Charpentier, 2013). Combined antiretroviral therapies (cART) have increased HIV-positive patients' life expectancy (Samji et al., 2013); however, HIV-associated neurocognitive disorders (HAND) have become more prevalent or remained at the same levels (Bingham et al., 2011; Clifford and Ances, 2013). HAND severity varies from deficiencies that do not affect daily living, asymptomatic neurocognitive impairment (ANI), to a more severe neurocognitive diagnosis known as HIV Associated Dementia (HAD) (Tozzi et al., 2003). Despite much effort to understand the causes of HAND, therapies targeting the specific mechanisms leading to the neuronal degeneration in HIV-infected brains are unavailable.
Multiple mechanisms of neurotoxicity appear to be at work among individuals with HAND, including HIV activation of apoptotic pathways (Kaul et al., 2001), dysregulation of calcium homeostasis (Lipton, 1994; Nath et al., 2000) and oxidative stress (Nath, 2002; Norman et al., 2008). Altered mitochondrial biogenesis (fission and fusion) is implicated in multiple neurodegnerative disorders, such as Alzheimer's and Parkinson's Disease (Deng et al., 2008; Wang et al., 2009, 2013). Mitochondrial fission is dependent upon the GTPase dynamin-related protein (DRP) 1, and is crucial for regulation of mitophagy as well as general mitochondrial distribution (Taguchi et al., 2007), allowing mitochondria-organelle-contacts, Ca + regulation and ATP supply to distant regions such as axonal and dendritic synapses in neurons (Berthet et al., 2014; Dickey and Strack, 2011; Li et al., 2004). DNM1L activity is regulated through nitrosylation and phosphorylation of serines: Ser-616 for activation, and Ser-637 for inactivation (Chang and Blackstone, 2007a, 2007b; Cho et al., 2009; Nakamura et al., 2010; Taguchi et al., 2007; Yan et al., 2015). Mitochondrial fusion requires two GTPase proteins, Mitofusin 1 and 2 (MFN) and optic atrophy (OPA) 1 (Chan, 2006; Chen and Chan, 2005). Mitochondrial fusion may serve as a stress response to protect cells from apoptotic cell death (Breckenridge et al., 2003) and excessive mitophagy (Gomes et al., 2011, 2012). Overactive mitochondrial fission is associated with apoptosis (Breckenridge et al., 2003), whereas mitochondrial fusion can mediate replenishment of mitochondrial proteins, DNA, and metabolic intermediates, and thus quality control (Twig et al., 2008). In healthy cells balance mitochondrial distribution and elongation, and therefore, an imbalance of fusion and/or fission protein activity may lead to dysfunction and cell death (Scorrano, 2013).
We recently reported that HIV-Tat promotes CDK5 localization to the cytoplasm of neurons and causes hyperphosphorylation of tau (Crews et al., 2007, 2011; Fields et al., 2015; Patrick et al., 2011). Interestingly, Duboff et al. showed that overexpression of tau leads to mitochondrial elongation and dysfunction via DNM1L mislocalization (DuBoff et al., 2012). Another study linked mitochondrial fusion and elongation with muscle degeneration that was rescued by MFN knockdown or DNM1L overexpression (Deng et al., 2008). Furthermore, HIV protein gp120 induces mitochondrial membrane permeablization and ultimatley leads to apoptosis (Ferri et al., 2000). Further delineating mechanisms by which HIV proteins cause mitochondrial dysfunction and subsequent neurotoxicity may reveal new opportunities for HAND therapies.
Here, we hypothesized that HIV proteins, released from infected cells of the central nervous system (CNS), interact with bystander neurons and cause alterations in mitochondrial fission/fusion processes directly, or as a result of HIV-protein induced mitochondrial dysfunction. To test this we assayed brain tissues from a well-characterized cohort of HIV + donors for expression of key mitochondrial biogenesis proteins. We complemented these studies with in vivo experiments using gp120 tg mice, and in vitro assays using SH-SY5Y neuronal cells and primary rat cortical neurons exposed to the HIV protein gp120. Overall, we observed changes in brain protein levels and mitochondrial morphology that suggest mitochondrial dynamics are shifted towards fusion and consolidation of the organelles in the neuronal soma of HAND donors. In vitro and in vivo studies suggest that increasing fission activity restores mitochondrial distribution throughout the neuron and neuronal viability.
4. Discussion
In the current study we report, for the first time, DNM1L and p-DNM1L-Ser616 protein levels are significantly reduced in brains of HAND donors compared to Non-HAND donors. These changes were reflected in degenerating neurons of HAND donors, and were associated with large and damaged mitochondria. Furthermore, GFAP-gp120 tg mouse brains had decreased DNM1L, increased MFN1, and elongated mitochondria with damaged cristae compared to Non-tg animals. In vitro, gp120 treatment reduced total and active DNM1L expression, increased MFN1, and reduced staining with potentiometric mitochondrial dye (Mitotracker Red) in neuronal cells. Gp120 treatment similarly reduced DNM1L levels in primary rat neurons, concomitant with increased capacity for extracellular acidification. Remarkably, DNM1L overexpression reversed neuroinflammation and neurodegeneration in the GFAP-gp120 tg mice and restored the intensity of Mitotracker staining to levels similar to control in primary rat neurons. Our results indicate that gp120 induced mitochondrial fusion, possibly, as a compensatory, but deleterious response that further exacerbates HIV-induced neuroinflammation and neurodegeneration. These findings are consistent with a recent study that showed DNM1L overexpression reverses HIV protein VPR-induced mitochondrial destruction (Huang et al., 2012).
Several neurodegenerative diseases are associated with abnormal mitochondrial dynamics, shown by altered regulation of mitochondrial fission and fusion proteins and mitochondrial distribution (Calkins et al., 2011; Tain et al., 2009; Zuchner et al., 2004). In Parkinson's disease, loss of parkin or PINK activity leads to impaired quality control by the fusion/fission machinery resulting in neuronal cell death (Tain et al., 2009). Decreased mitochondrial fission and fusion proteins have been reported in brains of Alzheimer's disease patients (Wang et al., 2009). Previous studies have shown that HIV derived proteins alter the mitochondrial caspase machinery, regulate calcium levels and alter glutamate toxicity leading to oxidative stress and apoptosis in neurons (Bonavia et al., 2001; Catani et al., 2000; Perfettini et al., 2005; Singh et al., 2005; Thomas et al., 2009). The data presented here are consistent with reports of altered mitochondrial homeostasis in neurodegenerative disorders, but are distinct in that increased mitochondrial fusion is the predominant observation. The question remains whether this fusion is the primary cause of, or a response to, neurodegeneration caused by HIV infection of the CNS. In either case, our data suggest excessive mitochondrial fusion in response to gp120 contributes to neurodegeneration. Understanding the mechanisms by which HIV associated proteins alter mitochondrial biogenesis and function may be crucial for understanding the causes of neurodegeneration in HIV infected patients.
We hypothesized that mitochondrial dynamics are altered in HAND cases compared to Non-HAND cases, due to the close association between mitochondrial morphology and metabolic functions. For instance high mitochondrial fragmentation occurs in cells that undergo apoptosis (Breckenridge et al., 2003), whereas mitochondrial fusion is crucial for sharing of mitochondrial contents and the rescue of damaged mitochondria (Twig et al., 2008). Mitochondria are typically in the post-fission state whereas fusion is a very selective process of quality control (Twig et al., 2008). TEM visualization of neuronal mitochondrial morphology revealed elongated and damaged mitochondria in HAND patients, which may suggest DNM1L is decreased and MFN1 is increased in an effort to salvage damaged mitochondria. Interestingly, and similar to our results, Twig et al. found that decrease of fission proteins resulted in an accumulation of damaged mitochondrial material and reduced respiratory function (Twig et al., 2008). Mitochondrial fission has been linked to autophagy and apoptosis, since damaged and depolarized mitochondria undergo mitophagy after fission and, as mentioned before, apoptosis can be induced by extensive mitochondrial fragmentation. It has been shown that increased fusion prevents autophagy and apoptosis, functioning as a rescue mechanism to transient cell stress (Chen and Chan, 2005; Gomes et al., 2011). Consequently the following question arises; should increased mitochondrial fusion in this case need to be interpreted as an accumulation of damaged mitochondrial components resulting from inefficient removal through mitophagy-coupled fission? Or does mitochondrial fusion serve as a buffering mechanism compensating damage induced by HIV proteins? The reduction in total and active forms of the mitochondrial fission protein DNM1L in both HAND and HIVE brains suggests that mitochondrial fission is either coordinately reduced by cell signaling to promote rescue of mitochondria and the cell, or this reduction is causing neurodegeneration through accumulation of otherwise mitophagy-bound organelles. It is also possible that both scenarios may be true and contribute to neurodegeneration. Glycogen synthase kinase (GSK) 3β has been shown to activate DNM1L GTPase activity (Yan et al., 2015). Conversely, cAMP-dependent protein kinase (PKA) attenuates DNM1L activity through phosphorylation (Chang and Blackstone, 2007a); hence, gp120 may induce alterations in these pathways that lead to the DNM1L expression observed in this cohort of HAND donors. It is note worthy that the donors classified as "Normal" had the highest CD4 counts of all donors and lower viral loads than donors with HAD (table). This may suggest that HIV replication, and hence gp120 level, is controlled in these patients, resulting in less neuronal damage. It will be important to determine which aspect of HIV infection is causing the neuronal mitochondrial damage in the CNS.
Animal models of HAND are valuable tools used to dissect the mechanisms by which HIV induces neurodegeneration in the CNS. The GFAP-gp120 tg mouse model is associated with behavioral deficits, neurodegeneration and neuroinflammation (Toggas et al., 1994). Multiple cellular pathways are disrupted in this model including autophagy and the CDK5 signaling pathway (Fields et al., 2013; Lee et al., 2013). Consistent with these reports, our data show that, specifically, mitochondrial cristae are malformed and mitochondria are elongated in damaged neurons in this model. Moreover, DNM1L and MFN1 expression levels are decreased and increased, respectively, suggesting gp120 is sufficient to induce these changes. Interestingly, a previous study reported that DNM1L heterozygote knockout mice had no synaptic or mitochondrial deficiencies (Manczak et al., 2012), and another showed that DNM1L inhibition attenuated neurotoxicity in mouse models of Parkinson's disease (Rappold et al., 2014). This may suggest that the impact of DNM1L levels is contextual. In the case of gp120 expression in the brain, gp120-induced mitochondrial toxicity may promote mitochondrial fusion to a deleterious level and thereby result in the pathology observed in the GFAP-gp120 tg mouse brains. Hence, it will be important to determine how gp120 directly causes these neuronal changes, and if changes in infected cells may also alter mitochondrial function in bystander neurons.
HIV can cause neuronal damage by inducing inflammation in infected microglia or bystander astrocytes or through direct interactions of HIV proteins with bystander neurons (Benos et al., 1994; Lipton, 1992; Walsh et al., 2014). Several HIV proteins lead to mitochondrial damage: transactivator of transcription (Tat) increases reactive oxygen species and alters mitochondrial membrane potentional (Nath et al., 2000). In addition, Viral Protein R (VPR) has been suggested to act on the mitochondrial permeability transition pore (MPTP) (Ferri et al., 2000; Jacotot et al., 2000), although the components of the pore as defined at the time have since been questioned (Bernardi and Di Lisa, 2015). Additionally, gp120 interacts with NMDA, chemokine receptors and lipid rafts on neurons leading to mitochondrial depolarization (Berth et al., 2015; Catani et al., 2000; Corasaniti et al., 1996). Endocytosed gp120 is trafficked throughout neuronal soma and axons, and may lead to direct neuronal damage (Bachis et al., 2006; Berth et al., 2015). Evidence is accumulating that HIV proteins, Tat and gp120, alter CDK5 signaling, which can lead to tau hyperphosphorylation, actin stabilization, and reduced DNM1L activity (Fields et al., 2015; Lee et al., 2013). In collaboration with Mocchetti's group, we recently reported that gp120 binds to TUBB3, a subunit of microtubules in neurons, and that this impedes mitochondrial trafficking and leads to increased mitochondrial size (submitted to Molecular Neurodegeneration). The current data are consistent with the increase in mitochondrial size. Interestingly, we show that gp120 decreased total and active DNM1L in human neuronal cells and primary rat neurons as early as 24 h after exposure. It is possible that the gp120 could activate the MPTP and induce mitochondrial swelling, and increasing DNM1L may facilitate recycling of these damaged mitochondria via mitophagy. Future studies on induction of MPTP in response to gp120 will be necessary to rule out this possibility. Nevertheless, these current data suggest that gp120 may cause the decreased DNM1L observed in HAND, HIVE and the GFAP gp120 tg mouse CNS neurons. Unlike in the neuroblastoma cell line, gp120 induced a reduction in MFN1 concomitantly with reduced DNM1L levels in primary rat neurons, suggesting that primary neurons may cease both mitochondrial fission and fusion in response to gp120. Increased ECAR by gp120-treated primary rat neurons suggests a rise in energy needs that is not being met by oxidative phosphorylation. Paradoxically, we observed no significant change, but an upward trend in maximal oxygen consumption rates that was highly variable. This may suggest that gp120 induces an energetic stress, and glycolytic capacity increases in order to meet neuronal ATP demands. Future studies will focus on the precise gp120-induced mechanisms at work in primary neurons. It will be particularly important to determine if gp120 affects turnover of mitochondria or mitochondrial proteins and if this can be modulated via DNM1L levels.
The notion that excessive mitochondrial fusion is induced by HIV and contributes to HAND is supported by our data showing that DNM1L gene delivery increased the Mitotracker staining of mitochondria and reduced accumulation of large perinuclear mitochondria in gp120-treated neuronal cells and primary rat neurons. Indeed, previous studies have shown that loss of mitochondrial fission depletes axonal mitochondria in neurons (Berthet et al., 2014). This is in contrast to a recent study that reported knockdown or chemical inhibition of DNM1L reduced gp120-induced pain sensation in the sciatic nerve of rats (Kanda et al., 2015). One possible, and simplistic explanation is that increased mitochondrial fission in the CNS of GFAP-gp120 tg mice helps to maintain neuronal function in an inflammatory environment that would otherwise minimize neuronal activity during response to the insult. In peripheral nerves, such as the sciatic, increased DNM1L may oversensitize the nerve in response to gp120, and thereby lead to the sensation of pain. The opposing gp120-induced responses of DNM1L in the CNS and the periphery are intriguing and deserve further investigation. DNM1L overexpression may reduce neuroinflammation and neurodegeneration in the GFAP-gp120 tg mouse model by maintaining distribution and quality control of mitochondria in neurons. This could be attributed to two DNM1L activities: 1) DNM1L may mediate increased mitophagy and clear damaged mitochondria; or 2) DNM1L may increase the number of mitochondria to a level that ensures proper distribution to active synapses and circumvents gp120 block of mitochondria trafficking on microtubules. Given that increased DNM1L appears to improve resistance to gp120-induced neurotoxicity, gp120 may increase fusion, through decreased DNM1L and increased MFN1, to a level that prevents proper neuron function. It will be interesting to see if blocking gp120 interaction with TUBB3 or altering cell signaling pathways in the GFAP-gp120 tg model will prevent neurodegeneration and behavioral deficits.
5. Conclusions
Overall, these data, along with the accompanying report, reveal a distinct alteration in mitochondrial morphology associated with HIV infection of the CNS and evidence that gp120 mediates these changes via altered mitochondrial fission/fusion and translocation along microtubules. The increase in mitochondria size is associated with severe HAND, and the alterations in fission/fusion machinery are observed early in neuronal models for HIV protein-mediated neurodegeneration. These data suggest that an imbalance in mitochondrial fission/fusion dynamics may be a compensatory mechanism that contributes to neurodegeneration in HAND patients. Furthermore, balancing DNM1L and MFN1 expression levels or activation status may rectify complications observed in GFAP-gp120 tg and other HAND mouse models with the end goal of treating HAND. Future studies will focus on the pathway through which gp120 induces alterations in mitochondrial fission/fusion dynamics.
2. Materials and methods
2.1. Study population
For the present study, we included a total of 27 HIV + cases categorized by HAND severity (normal, ANI, MND or HAD) (Table 1), from the California Neuro-Acquired immune deficiency syndrome (AIDS) Tissue Network (CNTN) at the University of California, San Diego. Cases had neuromedical and neuropsychological examinations within a median of 12 months before death. Most cases died as a result of acute bronchopneumonia or septicemia. Median postmortem interval was 12 h. Autopsy findings were consistent with AIDS and the associated pathology was most frequently due to systemic cytomegalovirus (CMV), Kaposi sarcoma, and liver disease. Subjects were excluded if they had a history of CNS opportunistic infections or Non-HIV-related developmental, neurologic, psychiatric, or metabolic conditions that might affect CNS functioning (e.g., loss of consciousness exceeding 30 min, psychosis, substance dependence). The diagnosis of HIV-encephalitis (HIVE) was based on the presence of microglial nodules, astrogliosis, HIV p24-positive cells, and myelin pallor. HAND diagnoses were determined from a comprehensive neuropsychological test battery administered according to standardized protocols (Woods et al., 2004).
3. Results
3.1. Clinical and neuropathological characteristics of HIV + donors
Before analyzing mitochondrial biogenesis protein levels and mitochondrial morphology we first meticulously collected and characterized clinical and neuropathological aspects of the HIV + cohort. For this purpose a total of 27 CNTN autopsy cases were analyzed in order to assess relevant differences between postmortem brain samples from HIV patients. We characterized the cohorts by brain weight, viral load (VL) and CD4 + cell count. The HAND cohort was predominantly male (88%) and the donors average age did not significantly vary with increasing HAND severity (Table 1). Plasma VL was similar among the groups, but CSF VL was highest in HAD donors (Table 1). Interestingly, the group with "normal" cognition had the highest CD4 counts, at 272.6 (Table 1). The "ANI" and "MND" groups had the lowest CD4 at 40.0 and 40.4, respectively, and the HAD CD4 count was in the 76.6 (Table 1).
3.2. Mitochondrial proteins are altered with increasing neuropsychological impairment and increasing neuropathology in brains of HIV + donors
Abnormal mitochondrial dynamics are associated with multiple neurodegenerative diseases, as shown by altered levels of mitochondrial fission and fusion proteins (Calkins et al., 2011; Tain et al., 2009; Zuchner et al., 2004). Additionally, previous studies have shown that HIV alters mitochondrial metabolic processes leading to oxidative stress and apoptosis (Parikh et al., 2015; Thomas et al., 2009). To determine the expression levels of mitochondrial fission/fusion in brains of HIV + donors, we analyzed frontal lobe lysates from HAND cases as well as Non-HAND cases (Table 1). Brain lysate membrane fractions were analyzed for DNM1L, p-DNM1L-Ser616, MFN1 and ACTB levels by immunoblot. In brain lysates from HAND cases, DNM1L and p-DNM1L-Ser616 protein band intensity decreased with increasing NCI, with the least being detected in HAD samples (Fig. 1A). Densitometry analyses of DNM1L and p-DNM1L-Ser616 bands show protein levels were decreased by 50% (Fig. 1B) and 40% (Fig. 1C), respectively, in brains from HAD cases compared to Non-HAND cases. DNM1L band signal were reduced in HIVE cases compared to HIV cases, while p-DNM1L-Ser616 bands were similar in HIV and HIVE brains (Fig. 1A). Densitometry analysis showed a 30% decrease in DNM1L levels (p < 0.05) (Fig. 1B) and a 20% increase in MFN1 levels (p < 0.05) (Fig. 1E) in postmortem brains of HIVE compared to HIV + patients.
3.3. Levels of DNM1L and MFN1 are altered and mitochondria are elongated in neurons of HAND patients
To determine if the biochemical alterations in DNM1L and MFN1 levels are associated with structural changes in neuronal mitochondria, we performed immunostaining for DNM1L, MFN1, and the mitochondrial transport protein TOMM40 in the frontal cortex of HIV + and HAND patients. DNM1L signal was reduced and showed a less punctate pattern throughout the soma of neurons in HAND patients compared to HIV + patients (Fig. 2A). Optical density analysis revealed a 35% decrease (p < 0.05) of DNM1L protein in neurons of HAND patients compared to Non-HAND patients (Fig. 2B). Conversely, MFN1 signal was increased in the neuronal soma from HAND patients (Fig. 2C). Measurement of signal intensity showed a 2-fold (p < 0.05) increase in MFN1 positive staining in neurons from HAND donors compared to HIV + (Fig. 2D). Signal for TOMM40 localized to large organelle structures in neuronal soma from HAND patients, suggesting increased mitochondrial size (Fig. 2E) Indeed, the average size of TOMM40 + structures were 80% larger in HAND neurons, compared to those of control HIV + donors (Fig. 2F).
Next, we aimed to determine if DNM1L localization to the mitochondrial membrane is reduced in brains of HAND donors compared to Non-HAND donors. Double-immunolabeling with DNM1L (green) and TOMM40 (red) was performed in frontal cortex sections from Non-HAND and HAND donors. DNM1L signal is robust and punctate in Non-HAND brains; however, DNM1L signal is less intense in HAND donors (Fig. 3A). TOMM40 signal is intense in both Non-HAND and HAND donors, but TOMM40 + punctae were larger in HAND tissues compared to Non-HAND (Fig. 3A). Merged images revealed a higher number of co-immunostained (yellow) puncta for the HIV + patient. Quantification analysis revealed 70% colocalization of TOMM40 and DNM1L for HIV + and only 25% for HAND (Fig. 3B), suggesting reduced DNM1L associated with mitochondrial membrane in HAND tissues.
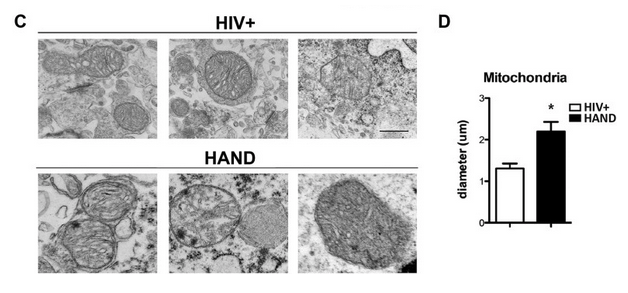
The ultrastructure of neuronal mitochondria reveals the size of individual mitochondria as well as the integrity of mitochondrial cristae, which can be indicative of mitochondrial health. Hence, we sought to investigate mitochondrial morphology in HIV + control and HAND utilizing transmission electron microscopy (TEM). Mitochondrial morphology was preserved in neurons from HIV + brains, with well-formed cristae and undisrupted mitochondria membranes (Fig. 3C). Conversely, mitochondria in HAND neurons were filled with discontinuous cristae, electron-dense structures and damaged double-membranes (Fig. 3D). Analysis of mitochondrial diameter revealed an increase of 80% (p < 0.05) in mitochondrial diameter in HAND brains compared to those from Non-HAND donors. These data suggest that decreased DNM1L in HAND leads to decreased mitochondrial fission and increased fusion.
3.4. DNM1L expression is decreased, MFN1 is increased and mitochondrial size is increased in gp120 tg mice
Animal models of HAND may provide insight to the mechanisms by which HIV proteins cause neuronal damage and lead to neuropathology and neurocognitive deficits. To investigate alterations in mitochondrial dynamics in an experimental model of HIV protein neurotoxicity, immunoblot analysis was performed on cortical extracts from gp120 tg and Non-tg mice. Lysate membrane fractions were immunoblotted for MFN1, DNM1L, and ACTB. DNM1L band signal was more intense and larger in Non-tg mouse brains compared to gp120 tg mice (Fig. 4A). The band corresponding to MFN1 was stronger in the gp120 tg mouse brains compared to those of Non-tg mice (Fig. 4A). Densitometry analysis showed DNM1L protein levels were decreased by 70% in gp120 tg mice (Fig. 4B), while MFN1 levels were increased by 120% in gp120 tg animals compared to Non-tg (Fig. 4C).
Additionally, we performed DNM1L and MFN1 immunostaining of vibratome cut sections from gp120 tg and Non-tg mouse brains. DNM1L staining appeared to be less intense in brain sections from gp120 tg mouse brains compared to Non-tg (Fig. 4D). Optical density analysis showed a 35% decrease (p < 0.05) in DNM1L signal in neurons of gp120 tg versus Non-tg animals (Fig. 4E). MFN1 signal is more intense and punctate in neurons in gp120 tg mice versus Non-tg mice (Fig. 4F). Quantification of signal intensity showed a 20% increase (p < 0.05) in MFN1 optical density in gp120 tg compared to Non-tg mice (Fig. 4G). TOMM40 signal was localized to large structures in neurons from gp120 tg mice (Fig. 4H). Using TOMM40 signal, mitochondrial average size was increased by 30% in neurons from gp120 tg compared to Non-tg mice (Fig. 4I).
Next, to determine if DNM1L localization to the mitochondrial membrane is reduced in neurons from gp120 tg mouse brains, we conducted double-immunolabeling of DNM1L (green) and TOMM40 (red). Confocal microscopy analysis displayed reduced quantity and intensity of DNM1L signal in neurons of gp120 tg versus Non-tg mouse brains (Fig. 5A). TOMM40 signal appeared to be more punctate and from larger structures in the gp120 tg mouse brains. (Fig. 5A). Overlap of DNM1L and TOMM40 signal revealed more merged puncta (yellow) in Non-tg compared to gp120 tg mice. Quantification of yellow pixel density demonstrated a 60% decrease in co-localization in gp120 tg compared to Non-tg animals (Fig. 5B).
Analysis of mitochondrial ultrastructure by TEM in gp120 tg and Non-tg animals was performed to corroborate mitochondrial damage observed in our human donor cohort. Neurons in gp120 tg mouse brains contained large and damaged mitochondria with electron dense regions and malformed cristae compared to Non-tg images (Fig. 5C). Diameter measurements revealed that mitochondrial size was significantly increased by 70% in gp120 tg mice (Fig. 5D). These results show that neuronal mitochondria are damaged and elongated in gp120 tg mouse brains.
3.5. Gp120, but not nef or Tat, decreases DNM1L, p-DNM1L-Ser616 and increases MFN1 in neuronal cells
HIV infection of the CNS causes a cascade of events in infected cells, as well as bystander cells. Mitochondrial alterations could be due directly to HIV proteins, or a downstream effect of inflammation or induction of intracellular signaling pathways. We first tested three HIV proteins-gp120, nef, and Tat for the ability to alter DNM1L levels in differentiated SH-SY5Y neuroblastoma cells. SH-SY5Y cells were treated with 10 or 100 ng/ml gp120, nef, or Tat for 24 h and then collected, lysed, fractionated and resolved by SDS-PAGE. Gp120 treatment decreased the DNM1L signal in a dose dependent manner, with the lightest band corresponding to the highest gp120 concentration (Fig. 6A). HIV nef or Tat had no observable effect on the DNM1L signal (Fig. 6B and C). Densitometry analysis of the DNM1L bands confirmed that gp120 decreases DNM1L protein levels by 40%, while nef or Tat had no significant effect on DNM1L levels (Fig. 6D-F). In order to determine the impact of gp120 on other fission/fusion proteins, we treated differentiated SH-SY5Y neuronal cells with increasing concentrations (10 and 100 ng/ml) of recombinant gp120 protein. Lysate membrane fractions were collected and immunoblotted for p-DNM1L-Ser616, MFN1, and ACTB. The band corresponding to p-DNM1L-Ser616 became more faint and thin with increasing doses of gp120 (Fig. 6G). The band corresponding to MFN1 became more intense and thick with increasing doses of gp120 (Fig. 6G). Densitometry analysis revealed a significant decrease and increase (p < 0.01) of p-DNM1L-Ser616 and MFN1 protein, respectively, at the highest concentration of gp120 (Fig. 6H and I). These data show that gp120 directly or indirectly decreased DNM1L and increases MFN1 levels in the particulate fraction of neuronal cells.
3.6. Gp120 increases mitochondrial size and decreases overall mitochondrial staining in neuronal cells; this effect is reversed by DNM1L overexpression and exacerbated by DNM1L knockdown
DNM1L mediates mitochondrial fission, and DNM1L activity may be induced by gp120 directly or as a compensatory mechanism in response to deleterious effects of gp120 on the neuronal cell. To determine if gp120 acutely causes mitochondrial morphology and activity alterations in our neuronal model, we infected SH-SY5Y cells with LV-DNM1L-HA (Fig. 7A) for 72 h and then treated with 100 ng/ml gp120 for 24 h, followed by incubation with Mitotracker Red. We then fixed and permeabilized the cells, immunostained for DNM1L and visualized mitochondria that were capable of sequestering the potentiometric Mitotracker Red. Infected cells were lysed and immunoblotted for DNM1L overexpression with anti-HA antibody; a strong band corresponding to DNM1L was detected in LV-DNM1L-HA-infected cells (Fig. 7B). LV-Control infected cells displayed evenly distributed mitochondria and DNM1L signal (green), but when treated with gp120 the Mitotracker staining was clearly accumulated in large structures with less intense red signal overall (Fig. 7C). DNM1L signal (green) was weakened in gp120-treated cells and less colocalized with Mitotracker signal (Fig. 7D). In LV-DNM1L infected cells, overall DNM1L immunostaining was increased along with Mitotracker signal (Fig. 5D and E). DNM1L and Mitotracker signal were equivalent in LV-DNM1L-infected cells treated with vehicle or gp120 (Fig. 7D and E). In LV-Control infected cells, gp120 caused a significant decrease in DNM1L (Fig. 7D) and Mitotracker signal (Fig. 7C); however, DNM1L overexpression rescued DNM1L (Fig. 7D) and Mitotracker signal (Fig. 7E). These data suggest increasing DNM1L expression and mitochondrial fission may counteract the negative effects that gp120 causes on neuronal cells.
As a proof of principal, we performed an experiment to knockdown DNM1L expression using siRNA specific for DNM1L. We transfected SH-SY5Y cells with control siRNA (siCON) or siRNA specific for DNM1L (siDNM1L) for 48 h and then treated with 100 ng/ml gp120 for 24 h, followed by incubation with Mitotracker Red. We then fixed and permeabilized the cells, immunostained for DNM1L and visualized mitochondria capable of accumulating Mitotracker Red. Transfected cells were lysed and immunoblotted for DNM1L overexpression; a strong band corresponding to DNM1L was detected in siCON transfected cells, but a weak band corresponding to DNM1L was detected in siDNM1L transfected cells (Fig. 8A). siCON transfected cells displayed evenly distributed mitochondria and DNM1L signal (green), but when treated with gp120 the Mitotracker staining was clearly accumulated in large structures with less red signal overall (Fig. 8B). DNM1L signal (green) was weakened in gp120-treated cells and less colocalized with Mitotracker signal (Fig. 8B). In siDNM1L infected cells, overall DNM1L signal was decreased along with Mitotracker signal (Fig. 8B). Quantification of DNM1L staining showed that gp120 reduced DNM1L signal by 25% in siCON transfected cells and by 50% in siDNM1L transfected cells (Fig. 8C). Quantification of Mitotracker staining showed that gp120 reduced DNM1L signal by 20% in siCON-transfected cells and by 65% in siDNM1L transfected cells (Fig. 8D). These data suggest reducing DNM1L expression and mitochondrial fission may worsen the negative effects that gp120 causes on neuronal cells.
3.7. Gp120 decreases DNM1L expression, increases mitochondrial size, increases H+ and decreases Mitotracker Red staining in primary rat neurons; this effect is reversed by DNM1L overexpression
Primary rat neurons serve as a model more relevant to human disease compared to cell lines such as SH-SY5Y. In order to determine the direct impact of gp120 on neurons, we treated primary rat neurons with increasing concentrations (10 and 100 ng/ml) of recombinant gp120 protein. Lysate membrane fractions were collected and immunoblotted for DNM1L, MFN1, and ACTB. The band corresponding to DNM1L became more faint and thin with increasing doses of gp120 (Fig. 9A). Interestingly, the band corresponding to MFN1 was also reduced in size and density with increasing doses of gp120 (Fig. 9B). Densitometry analysis revealed a significant decrease (p < 0.01) of DNM1L and MFN1 protein, respectively, at the highest concentration (Fig. 9C). These data show that gp120 directly or indirectly decreased DNM1L and MFN1 levels in the particulate fraction of primary rat neurons.
Mitochondria produce ATP through oxidative phosphorylation via the ETC; however, when oxygen supplies are short or the ETC is not functioning properly, glycolysis compensates for the lack of ATP. Lactate is a by-product of glycolysis, and hence, glycolytic activity may be indirectly measured through ECAR (H+ production rate). Indeed, measurements of extracellular pH have long been used as an indicator of glycolytic activity in cultured cells (Balaban and Bader, 1984; Divakaruni et al., 2014). To determine if neurons increase ECAR in response to gp120, primary rat neurons were treated with gp120 (100 ng/ml) for 24 h and then ECAR and respiration was measured on a Seahorse extracellular flux analyzer. The maximal ECAR following addition of oligomycin (when oxidative phosphorylation is blocked) increased in response gp120 ECAR by 23% (p < 0.01; Fig. 9D). Under these conditions, the maximal uncoupler-stimulated respiratory rates did not change significantly. These data suggest that treatment with gp120 enhances the glycolytic capacity of primary cortical neurons.
To determine if gp120 acutely causes alterations in mitochondrial morphology and staining with a potentiometric dye in primary rat neurons, we infected primary cultures with LV-DNM1L-HA for 72 h and then treated with 100 ng/ml gp120 for 24 h, followed by incubation with Mitotracker Red. We then fixed and permeabilized the cells, immunostained for DNM1L and visualized active mitochondria for Mitotracker signal. Infected cells were lysed and immunoblotted for DNM1L overexpression with anti-HA antibody; a strong band corresponding to DNM1L was detected in LV-DNM1L-HA-infected cells (Fig. 9E). LV-Control infected cells displayed evenly distributed mitochondria and DNM1L signal (green), but when treated with gp120 the Mitotracker signal was clearly accumulated in large structures with less Mitotracker signal overall (Fig. 9F). In LV-Control infected cells, DNM1L signal (green) was weakened (p < 0.05) by gp120-treatment (Fig. 9G). In LV-DNM1L infected cells, overall DNM1L signal was increased along with and DNM1L signal remained high (p < 0.01) following gp120 treatment (Fig. 9F). In LV-Control infected cells, Mitotracker signal (red) was less intense (p < 0.05) after gp120-treatment (Fig. 9G); however, DNM1L overexpression restored Mitotracker signal in gp120-treated cells (Fig. 9H). These data suggest increasing DNM1L expression and mitochondrial fission may counteract the negative effects that gp120 causes on neuronal cells.
3.8. LV-mediated DNM1L overexpression reduces mitochondrial size, neurodegeneration and neuroinflammation in GFAP-gp120 tg mouse brains
Given the study results on neuronal cell lines and primary rat neurons showing a protective effect of DNM1L against gp120, we next sought to determine if increasing DNM1L levels in GFAP-gp120 tg mouse brains reduces the neuropathology associated with this NeuroAIDS model. We stereotaxically infected LV-DNM1L-HA in to the brain. Two weeks post-injection, mice were euthanized and brains were processed for immunohistochemical analysis. Non-tg and GFAP-gp120-tg mouse brains that were infected with LV-Con showed no signal when immunostained for the HA epitope; however, the signal was intense around the site of injection in brains of the Non-tg and GFAP-gp120-tg mice infected with LV-DNM1L-HA (Fig. 10A). Optical density for HA immunostaining was increased 17-fold in mice infected with LV-DNM1L-HA compared to the mice infected with LV-Con (Fig. 10B). To determine the localization and size of mitochondria, we immunostained vibratome sections of the mouse brains with antibody for TOMM40 (green). TOMM40 signal is punctate and evenly distributed throughout the image in Non-tg mouse brains (Fig. 10C). TOMM40 signal is associated with large and less numerous organelles in the GFAP-gp120 tg mouse brains that were infected with LV-con; however, the TOMM40 signal, in GFAP-gp120 tg mouse brains infected with LV-DNM1L-HA, is more dispersed, numerous and associated with visibly smaller organelles (Fig. 10C). Analysis of the average size of the TOMM40 puncta shows that mitochondria are reduced by 20% in Non-tg mouse brains infected with LV-DNM1L-HA (Fig. 10D). Conversely, TOMM40 + puncta are 50% larger in GFAP-gp120 tg mouse brains compared to the TOMM40 + puncta in corresponding Non-tag brains (Fig. 10D). These data suggest that gp120-induced reduction in DNM1L levels and activity leads to increased mitochondria size and consolidation of the organelles.
To determine if DNM1L overexpression, via lentiviral vector infections, reduces neuronal mitochondrial size and/or neuropathology associated with the GFAP-gp120 tg model, we immunostained vibratome sections with GFAP and MAP2 antibodies as indicators of astrogliosis and neurodegeneration. GFAP signal is low throughout brain tissues of Non-tg mice, both LV-con and LV-DNM1L-HA infected (Fig. 11A). Conversely, GFAP signal is intense and more abundant in the GFAP-gp120 tg mouse brains, as previously reported (Fig. 11A) (Toggas et al., 1994). GFAP-gp120 tg mice infected with LV-DNM1L-HA presented GFAP staining much less intense and less dispersed compared to GFAP-gp120 tg mice infected with LV-con (Fig. 11A). Optically density analysis of GFAP signal shows the astrogliosis is similar in both groups of Non-tg mouse brains; however, LV-DNM1L-HA infusion reduced GFAP signal by 30% in the GFAP-gp120-tg mouse brains (Fig. 11B).
Next, we performed immunostaining of brain tissues with MAP2 to analyze neuronal integrity in the Non-tg and GFAP-gp120 tg mice. MAP2 signal (green) is uniformly staining pyramidal neurons throughout brain tissues of Non-tg mice, both LV-con and LV-DNM1L-HA infected (Fig. 11C). Conversely, MAP2 signal is reduced and more sparse in the GFAP-gp120 tg mouse brains, as previously reported (Toggas et al., 1994) (Fig. 11C). Remarkably, GFAP-gp120-tg mice infected with LV-DNM1L-HA presented MAP2 signal in similar intensity as the Non-tg mouse brains (Fig. 11C). Optically density analysis of the area of the neutrophil staining for MAP2 shows equivalent signal in both groups of Non-tg mouse brains; however, MAP2 signal is reduced by 20% in GFAP-gp120 tg mouse brains that were infected with LV-con compared to Non-tg mouse brains (Fig. 11D). DNM1L overexpression increases MAP2 signal area by 15% in GFAP-gp120 tg mouse brains, compared to those infected with LV-con (Fig. 11D). These data suggest that maintaining mitochondrial fission in GFAP-gp120 tg mouse brains may reduce neuroinflammation and neuroinflammation.
|
| | | | | | |
|