 |
|
|
| |
Determinants of fibrosis progression and regression in NASH
|
| |
| |
Download the PDF here
Jnl of Hepatology Jan 2018 - Detlef Schuppan1,2, Rambabu Surabattula1, Xiao Yu Wang1 1Institute of Translational Immunology and Research Center for Immunotherapy, University of Mainz Medical Center, Mainz, Germany; 2Division of Gastroenterology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, USA
Summary
Cirrhosis has become the major liver-related clinical endpoint in non-alcoholic steatohepatitis (NASH). However, progression to cirrhosis is less predictable in NASH than in other chronic liver diseases. This is due to the complex and multifactorial aetiology of NASH, which is determined by lifestyle and nutrition, multiple genetic and epigenetic factors, and a prominent role of hepatic and extrahepatic comorbidities. Thus, modest changes in these cofactors can also induce fibrosis regression, at least in patients with precirrhotic liver disease. Fibrogenesis in NASH correlates with, but is indirectly coupled to, classical inflammation, since fibrosis progression is driven by repetitive periods of repair. While hepatocyte lipoapoptosis is a key driving force of fibrosis progression, activated hepatic stellate cells, myofibroblasts, cholangiocytes, macrophages and components of the pathological extracellular matrix are major fibrogenic effectors and thus pharmacological targets for therapies aimed at inhibition of fibrosis progression or induction of fibrosis reversal. The advent of novel, highly sensitive and specific serum biomarkers and imaging methods to assess the dynamics of liver fibrosis in NASH will improve detection, stratification and follow-up of patients with progressive NASH . These non-invasive tools will also promote the clinical development of antifibrotic drugs, by permitting the design of lean proof-of-concept studies, and enabling development of a personalised antifibrotic therapy for patients with rapid fibrosis progression or advanced disease.
Relevance of liver fibrosis in NASH
NASH is distinct from other liver diseases, because it is tightly associated with comorbidities of the metabolic syndrome, such as insulin resistance and type 2 diabetes, and cardiovascular complications linked to hypertension and dyslipidaemia. These comorbidities already exist in mere NAFLD, which is defined by the absence of fibrosis and major liver inflammation.[1], [2], [3], [4] These non-hepatic companion diseases represent the major comorbidities and causes of mortality in NAFLD and the early stages of fibrotic NASH, up to stage 2 fibrosis. Liver-related morbidity and mortality only increase significantly beyond stage 1 fibrosis, especially with the emergence of cirrhosis. Thus, prevention and therapy must address two largely independent targets: the metabolic complications which are treatable with a range of medications, and moderate to advanced fibrosis (stage 2-4) for which no approved drugs exist. To prevent liver-related mortality, the primary goal is to reverse advanced fibrosis, or prevent progression to cirrhosis in patients that can be identified as rapid progressors. Whereas antifibrotic treatment of NASH patients with stage 1 fibrosis is less meaningful, as mild fibrosis may not progress and can even regress with minor lifestyle changes or pharmacological treatment of the metabolic syndrome. Stratifying patients with NASH into those with mild vs. advanced fibrosis is supported by several smaller studies and a recent meta-analysis that demonstrated a 9.57-, 16.69- and 42.3-fold increase in liver-related mortality, in subjects with stage 2, 3 and 4 fibrosis vs. subjects with no (stage 0) fibrosis, with only a 1.41-fold mortality increase in patients with stage 1 fibrosis.5 Notably, all-cause mortality, which is dominated by cardiovascular complications in early stages, was increased to a similar level, i.e. 2-3-fold, both in early stages and advanced stage 3-4 fibrosis. A large single retrospective study included 619 US patients with biopsy confirmed NASH who were followed up for an average of 12.6 years.6 A total of 33.2% reached the hard endpoints of all-cause mortality or liver transplantation. Fibrosis of any stage vs. no fibrosis was associated with a hazard ratio (HR) for these endpoints of 1.86, increasing to 3.8 and 10.9 for stage 3 and 4 fibrosis, while the HR for liver-related hard endpoints rose to 14.2 and 51.5, respectively. The only other relevant predictors were age (HR 1.07), type 2 diabetes (HR 1.62) and smoking (HR 2.62). Interestingly, statin use decreased the risk of mortality or transplantation significantly (HR 0.32). The relevance of advanced fibrosis as the primary hepatic endpoint in clinical studies has now been recognised by the regulatory authorities and drug developers. Advanced fibrosis has become a central focus in the ongoing phase III and in most phase II studies,7 with an increasing demand to include compensated cirrhotics in current clinical trials.8
Key Point
Prevention and therapy of NAFLD must address two largely independent targets: the metabolic complications, and moderate to advanced fibrosis
Epidemiology and natural history of liver fibrosis in NASH
There is no doubt that the epidemic of NAFLD and NASH is mainly caused by overnutrition, unhealthy food constituents and a sedentary lifestyle, which have steadily and inconspicuously infiltrated our daily lives. From 1994-2015, obesity, as defined by a body mass index (BMI) >30 in Western and >28 in far Eastern countries, has increased roughly twofold in the US. This is perfectly paralleled by the prevalence of type 2 diabetes, which reaches 10% or more in some Southern and Midwestern states of the US[9], [10] and in more affluent North African and Middle Eastern countries.11The reported national and regional prevalence of NAFLD and the overall prevalence of NASH-related cirrhosis largely parallel these developments in adults, as well as in children and adolescents.[12], [13], [14], [15]
While reliable data on obesity and type 2 diabetes can be obtained on a nationwide scale, the clear diagnosis of NAFLD and especially the differentiation between NAFLD and NASH in large representative cohorts is difficult. This is because the only accepted diagnostic criterion is liver biopsy, with specified criteria for significant hepatic steatosis, hepatocyte ballooning and inflammation (NAS score >4 or >5).[16], [17]
Studies based on follow-up biopsies in patients with non-inflammatory fatty liver (NAFL) and NASH revealed interesting characteristics of fibrosis progression vs. regression in patients under standard follow-up. In a study from Newcastle that included 108 patients who were followed up for an average of 6.6 years with a second biopsy, 42% progressed, 40% remained stable, and 18% regressed.18 Interestingly, 10 of the 27 NAFL patients with stage 0 fibrosis progressed, six of whom developed stage 3 fibrosis. The best explanation for spontaneous fibrosis regression, besides biopsy sampling variability, may be favourable lifestyle changes. Another study from France followed up 70 patients with untreated NAFLD using a second biopsy after a mean of 3.7 years. A total of 25 patients had NAFL and 45 had NASH and/or advanced fibrosis. Of the patients with NAFL, 16 developed NASH, eight with ballooning and six with bridging fibrosis (stage 3) on follow-up. Patients with disease progression were older and had worsening of their metabolic risk factors. Interestingly, ballooning and bridging fibrosis correlated with a reduction in alanine aminotransferase (ALT), more weight gain, and a higher incidence of diabetes during follow-up.19 In the same cohort, mere fatty liver was an independent predictor of non-liver morbidity, such as carotid artery stenosis.20 A meta-analysis of 2015 has summarised the paired biopsy studies.21 Taken together, fibrosis in NASH can regress spontaneously in earlier stages, whereas reversibility decreases from stage 3 on. Mere fatty liver can progress to advanced fibrosis, indicating that even the abundant subjects with NAFL would need regular monitoring for fibrosis progression.
Overweight and type 2 diabetes as key determinants of fibrosis progression and regression
A recent extensive meta-analysis has assessed the global burden of NAFLD and NASH, and disease outcome, demonstrating a prevalence of NAFLD between 15% and 30% in most affluent and developing countries.22 To clearly identify individuals with NAFLD that have significant liver inflammation and fibrosis would completely overwhelm any healthcare system, since we do not currently have a simple and cheap non-invasive tool with sufficient sensitivity and specificity. This problem is further potentiated by expected increases in NAFLD disease burden in the next few years.23
Key point
The natural history of NASH is less predictable than for other chronic liver diseases, but the important role of lifestyle changes, including weight reduction, physical exercise and nutrient composition is becoming clear.
Notably, the natural history of NASH is much less predictable than that of other chronic liver diseases, such as viral hepatitis B or C, where progression is tightly linked to the underlying cause, and where causal treatment like efficient viral suppression or elimination halts fibrosis progression and even causes reversal.[24], [25] The important role of lifestyle changes, especially weight reduction, but also physical exercise, and a healthier, yet ill-defined, nutrient composition on inflammation and fibrosis in NASH is highlighted in a Cuban-US study. It demonstrated that an intensive weight loss programme in 293 patients with NASH, 261 of whom were biopsied after 52 weeks, led to resolution of NASH (NAS score below 3) in 25%, reduced the NAS score in 47% and reduced fibrosis in 19%. In those patients who lost ≥10% of body weight, NASH resolved in 90% and fibrosis improved in 45%.26 Based on these data, the authors developed an index, based on age, pre-intervention type 2 diabetes and NAS score ≥5, weight loss and ALT normalisation, with a high predictive value for NASH resolution (area under receiver operator curve <5).27 Moreover, follow-up studies on morbidly obese patients that underwent bariatric surgery have demonstrated that the extent of weight loss correlates with the degree of resolution of NASH and even fibrosis regression. Thus, a prospective study from France, including 109 subjects followed up for one year, showed that patients who underwent laparoscopic gastric banding lost less weight than those who underwent gastric bypass (change in BMI 6.4 vs. 14.0, respectively) which lead to less resolution of persistent NASH (69.6% vs. 92.4%, respectively).28 Notably, the overall NAS score was reduced from five to one, and fibrosis regressed in 33.8% of cases. These findings also stress the point that patients with fibrotic NASH need to be monitored frequently for lifestyle changes and disease activity, a task which would ideally be done with reliable serological tests to assess hepatic fibrogenesis and fibrolysis, as outlined below.
The role of intestinal microbiota
While studies in mice show that using defined intestinal microbial colonisations can strikingly influence the metabolic phenotype, including obesity, insulin resistance and features of NASH,[29], [30] the role of the intestinal microbiota in patients is less clear. Clinical studies that establish correlations with disease severity are just beginning to emerge.[31], [32] Undoubtedly intestinal microbes determine the efficiency of nutrient usage and thus caloric uptake, affecting weight gain and insulin sensitivity.33 This was nicely shown in an interventional study using faecal transplantation between lean subjects and patients with obesity and insulin resistance.34 Beneficial effects of bowel decontamination by antibiotics have been observed in patients with cirrhosis, but these are mainly due to intestinal barrier defects in advanced liver disease, particularly alcoholic cirrhosis, that permit bacterial translocation and activation of proinflammatory toll-like and other innate immune receptors in the liver, mainly on resident or recruited macrophages and Kupffer cells.35 However, in a small uncontrolled study of 15 patients with biopsy proven noncirrhotic NASH, treatment with rifaximin for six weeks did not improve ALT, steatosis and insulin resistance.36 At present, an effect of microbiota on fibrosis progression or regression remains elusive. Therefore, more clinical research beyond mouse models and mere correlations must be conducted, including interventional studies, to gauge the relevance of intestinal microbiota and their metabolome, including bile acid metabolism, as prime drivers of NASH pathogenesis.[37], [38]
Biopsy and other predictors of fibrosis and fibrosis progression
Liver biopsy has remained the gold standard for staging and grading of NASH, for follow-up and natural history studies, and for the monitoring of antifibrotic drug effects in clinical trials. However, the massive number of potential patients with NASH, an estimated 15-20% of those with fatty liver, precludes liver biopsy for case finding. Moreover, liver biopsy is fraught with a high sampling variability because of the small size of the acquired tissue sample. Thus, well controlled studies have shown a one stage difference (out of a possible five stages) between two large liver biopsies taken adjacently or from the left and right lobes, in 40% of patients with NASH.[39], [40] Therefore, current phase IIB clinical studies require a high number of well stratified patients (>200) and a long duration of treatment (>1.5 years) to identify drugs or regimens with a significant antifibrotic potential. For phase IIA proof-of-concept trials, smaller numbers of patients and short-term surrogate endpoints, such as ALT and fat content, are currently used. As outlined below, these trials increasingly incorporate other non-invasive markers that assess fibrosis, such as magnetic resonance elastography and serum fibrosis and fibrogenesis markers.
Histology may be refined to decrease sampling variability, by quantification of α-smooth muscle actin positive, fibrogenic, hepatic stellate cells (HSCs) and myofibroblasts (MFs), and by morphometrical collagen determination after Sirius red staining.[25], [41], [42] The recent development of dual photon imaging on unstained formalin-fixed sections may offer a more refined assessment of deposited collagen and collagen architectural changes, including fibril diameter, length and crosslinking.43 A novel technology for quantifying hepatic fibrogenesis has been developed, based on the daily ingestion of deuterated water for 3-5 weeks to label newly synthesised liver collagen, followed by measurement of the deuterated collagen fraction from a liver biopsy via mass spectrometry.44 In a well-characterised cohort of 21 patients with NAFLD, hepatic collagen synthesis rate correlated with biopsy-monitored fibrosis progression.44 Moreover, deuterated plasma lumican, a proteoglycan of the fibrotic matrix, correlated with the collagen synthesis ratio.44 Despite its complexity and invasiveness, this method may be useful to validate other, more easily determined non-invasive markers of fibrogenesis.
Imaging and non-invasive markers of fibrosis, fibrogenesis and fibrolysis
Key point
Much effort is being invested in the search and validation of serum markers that predict fibrosis stage and the dynamics of fibrogenesis and fibrolysis.
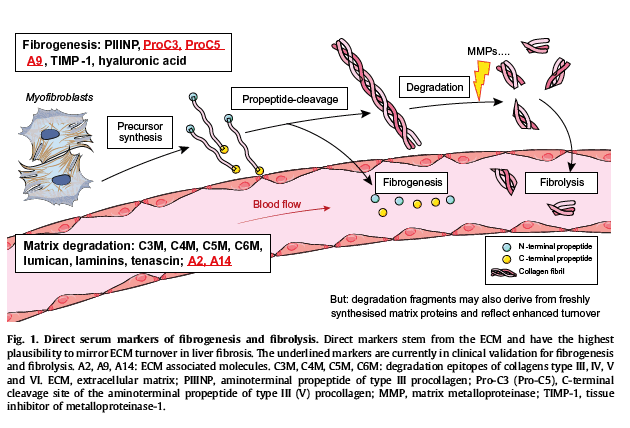
Histological staging is clearly needed to guide the development, validation and improvement of non-invasive imaging to assess fibrosis, such as ultrasound and magnetic resonance elastography.[45], [46] These non-invasive tools are not likely to be sensitive enough to identify minor changes in fibrosis, e.g. within a few weeks or months after lifestyle changes or specific anti-inflammatory or antifibrotic therapy. Recently, preclinical developments, such as imaging of elastin or collagen probes,[47], [48] and even of fibrogenesis by targeting the integrin αvβ6,49 hold promise for a reliable assessment of fibrosis and its dynamics over the whole liver. Moreover, much effort is invested in the search and validation of serum markers that predict the stage of fibrosis,[45], [46], [50], [51], [52], [53] and, more importantly, the dynamics of fibrogenesis and fibrolysis.[52], [53], [54], [55], [56], [57] True markers of fibrogenesis would enable the selection of patients likely to respond to antifibrogenic therapies, i.e. those with significant fibrogenic activity, and to detect responders to this therapy. Fibrolysis markers would assess the response to treatments that remove excess fibrotic tissue, both in those with active and those with inactive fibrogenesis but advanced fibrosis or cirrhosis. It is expected that in the near future their combination will permit assessment of the net effect of an antifibrotic therapy a few days or weeks after the initiation of therapy, and on a regular basis, e.g. to mirror the progression and regression rate and predict the final outcome, be it histological or clinical. Such dynamic markers are currently developed within the European consortia EPOS (European Project on Steatohepatitis) and LITMUS (Liver Investigation: Testing Marker Utility in Steatohepatitis). They are largely "direct markers" derived from molecules that are involved in extracellular matrix (ECM) build up or removal. The rationale behind these markers and the most promising single tests for fibrogenesis and fibrolysis are shown (Fig. 1). One of the novel markers developed and validated in the EPOS cohort, Pro-C3, a direct marker of collagen synthesis, is increasingly used in proof-of-concept clinical studies to predict the antifibrotic potential of novel agents for the treatment of NASH.57
Inflammation and the dynamics of fibrosis progression
Liver fibrosis, like fibrosis in general, is the result of a protracted wound healing process.[8], [52], [53], [58], [59], [60], [61], [62], [63] We hypothesise that ongoing fibrogenesis, namely the active accumulation of scar tissue (ECM), does not follow a simple path of progression from steatosis to steatohepatitis to cirrhosis. Rather, based on preclinical data in optimised murine models of non-alcoholic steatohepatitis (NASH),[64], [65]progression appears to result from repetitive bouts of inflammation alternating with an anti-inflammatory, reparative immune response. This is particularly evident in a disease where environmental and lifestyle changes can quickly affect the course of inflammation and fibrosis.[3], [26] As detailed later, short-term, classical proinflammatory processes (type 1 inflammation, represented by T helper (Th) 1 cells and M1 macrophages) are associated with enhanced tissue destruction and fibrolysis, i.e. ECM degradation and removal. In contrast, the ensuing anti-inflammatory immune response (type 2 inflammation, represented by Th2 T cells and M2 macrophages) is largely profibrotic, especially when repetitive phases of type 1 inflammation are followed by type 2 inflammation.66 Following this paradigm, the simple view of correlating classical M1 inflammation with fibrogenesis does not hold true. It is rather the extent and length of the reparative relative to the inflammatory periods that may finally determine the speed of fibrosis progression (Fig. 2). In NASH this hypothesis also explains why histological and surrogate serum markers of inflammation, such as ALT, show an overall correlation with the risk of fibrosis progression in larger cohorts, but are unable to predict progression on an individual basis. In the same vein, effective anti-inflammatory therapies do not necessarily predict antifibrotic effects, nor should drugs that show no anti-inflammatory effects be discarded as ineffective antifibrotics, especially when their mechanism of action suggests such activity. A good example is the promising antifibrotic effect of the dual chemokine receptor (CCR)-2 and -5 antagonist, cenicriviroc, in the absence of the expected anti-inflammatory activity.67Therefore, for market approval of antifibrotic drugs in NASH (and other fibrotic liver diseases) there is currently no proven alternative to follow-up histology and fibrosis staging, as a primary endpoint. However, this is quickly changing because of emerging technologies for quantitative imaging of fibrosis and its dynamics, and the ongoing development of serum surrogate markers of fibrogenesis and fibrolysis (Fig. 1).
Mechanisms of liver fibrosis progression in NASH
While pharmacological therapies for the complications of the metabolic syndrome are numerous, therapies for liver inflammation in NASH are just emerging, and approved therapies for liver fibrosis remain elusive.[8], [52], [53], [58], [59], [60], [61], [62], [63] A better understanding of NASH pathogenesis, including the fibrogenic and inflammatory drivers of disease progression is crucial. Undoubtedly, nutrient caloric overload, which is not matched by nutrient expense, leads to an overflow of peripheral and liver fat stores. The ensuing adipose tissue and liver inflammation represent the first hit in NASH pathogenesis. Under such circumstances, the liver, peripheral adipose tissue and intestine interact via cytokine, growth factor and adipokine secretion, with the liver taking centre stage in metabolic regulation.[1], [2], [37] Hepatocytes are major metabolic and lipid handling cells. During lipid overflow in hepatocytes, lipids, particularly free fatty acids derived from the periphery, escape safe storage as macrovesicular fat. Under conditions of mitochondrial overstraining, this leads to mitochondrial and peroxisomal dysfunction and enhanced oxidative stress. Uncontrolled and incomplete lipid oxidation generates toxic lipid products which cause hepatocyte damage and finally lipoapoptosis.[68], [69], [70] Lipoapoptosis in hepatocytes can be considered a prime cause of liver inflammation in NASH, by attracting and activating inflammatory cells. When this inflammatory process becomes chronic, further metabolic deterioration and fibrosis will ensue. Many novel approaches to causally treat these metabolic derangements appear to affect liver fibrosis, reviewed previously and again in this issue.[71], [72]
The liver extracellular matrix and the major scar tissue producing cells
Key point
Collagens are the most abundant ECM components in fibrosis and are significantly increased in cirrhosis.
The bulk of ECM components in scar tissue are produced by activated MFs, which either derive from activated HSCs or from activated (portal and perivascular) fibroblasts.[8], [52], [53], [58], [59], [60], [61], [62], [63] Therefore, these cells represent direct downstream target cells for therapeutic intervention. Fibrosis, defined by the excessive accumulation of ECM, goes hand in hand with altered angiogenesis, and eventually leads to the severe architectural changes of cirrhosis.[41], [73], [74] Collagens are the most abundant ECM components in fibrosis, increasing up to tenfold in cirrhosis.[74], [75], [76] Collagen type I has recently been shown to be a highly promising target for specific pharmacological intervention using clinical grade siRNA technologies in vivo.[77], [78] In the same vein, induced genetic ablation of the procollagen α1(I) gene during active fibrogenesis not only reduced overall collagen accumulation by 50%, but also attenuated liver inflammation, which implicates excess collagen type I as driver of inflammation and suggests that a (pro-)collagen type I targeted therapeutic approach is safe.79 There are also numerous other ECM molecules that are either indicators or therapeutic targets of liver fibrosis.[54], [74] Importantly, absolute and relative collagen composition changes dramatically with advancing liver fibrosis, with some pathological species becoming predominant, potentially serving as novel targets for specific pharmacological therapies.[54], [74], [76] Moreover, all liver cells receive positional and differentiation signals from these normal or pathological ECM molecules, mainly via specific integrin or proteoglycan receptors. Therefore, inhibition of certain integrins or receptor proteoglycans is another promising field for developing specific antifibrotics.[53], [80], [81], [82], [83], [84], [85], [86]
An approach targeting the extensive crosslinking of major ECM proteins, such as collagens and elastin, by the enzyme class of lysyl oxidases (Lox), especially lysyl oxidase like 2 (Loxl-2), is attractive, since a less crosslinked ECM should be more susceptible to proteolytic degradation. Based on a preliminary experimental study,87 a humanised blocking antibody to Loxl-2 (Simtuzumab) was tested in several hundred patients with early and late stage NASH. However, as before in pulmonary fibrosis,88 no significant antifibrotic activity was shown in NASH,89 which has dampened enthusiasm for this therapeutic target. These disappointing results may have been due to both limited penetration of the antibody into human scar tissue and the antibodies' relative inefficiency for neutralising the target, since preclinical data indicate that Loxl-2 and general Lox-inhibition effectively prevent liver fibrosis progression and facilitate its reversal, including a protection from hepatic neoplasia.[90], [91], [92] Therefore, efforts to develop different Lox and Loxl2 inhibitors are justified.
The role of multiple hits in fibrosis progression
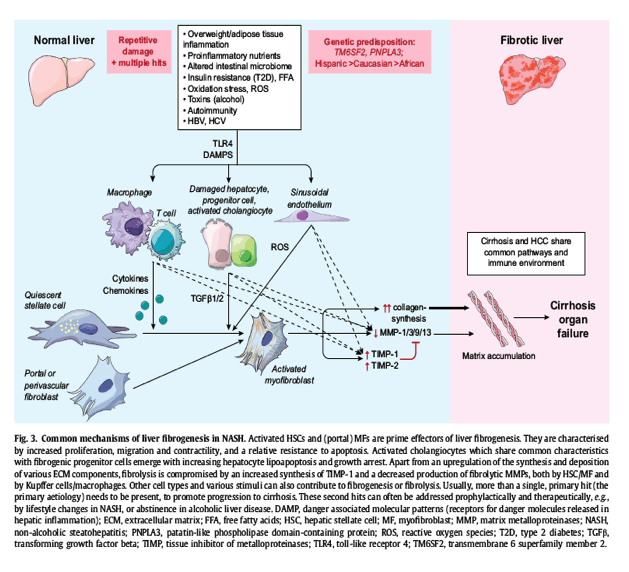
Apart from hepatocyte lipoapoptosis, a variety of other stimuli, such as toxins, viruses, or cholestasis contribute to fibrogenesis in NASH, either indirectly or directly. The presence of multiple hits, the hierarchy of which remains unclear,93 affects the inflammatory and fibrogenic phenotype. Their identification is key to an appropriate stratification of patients for clinical studies. In addition, genetic and emerging epigenetic predispositions are important determinants of metabolic, inflammatory and fibrotic NASH, as well as alcoholic liver disease.[94], [95], [96] The contribution of these hits to the cellular machinery of fibrogenesis is illustrated (Fig. 3). Thus, apart from their genetic predisposition, patients with rapid fibrosis progression usually have several hits, such as alcohol abuse, and other often unidentified exposures to toxins, detrimental nutrients or drugs, HCV infection or autoimmune liver diseases. Therefore, the elimination or appropriate treatment of these contributing hits will alleviate fibrosis progression and decrease the risk of development of cirrhosis and HCC.\
Key point
Identifying the multiple hits that underlie the development of fibrosis are key for stratifying patients for clinical studies.
Major genetic determinants that affect NASH progression
Genetic polymorphisms in PNPLA3 (patatin-like phospholipase domain-containing protein) and TM6SF2 (transmembrane 6 superfamily member 2) are the best validated determinants of NASH progression. The isoleucin to methionine polymorphism of PNPLA3 (I148M) is associated with an increased risk of advanced fibrosis, including in patients with a variety of other liver diseases, and is an independent risk factor for HCC in patients with NASH or alcohol-related cirrhosis.[97], [98] I148M leads to a loss of function, resulting in increased hepatocyte triglyceride accumulation. The exact molecular mechanisms leading to hepatic fibrosis and carcinogenesis remain unclear. TM6SF2 is a putative master regulator of metabolism that not only determines the risk of advanced liver disease in NASH, but also cardiovascular disease outcomes. Notably, the TM6SF2 polymorphisms that predict NASH progression, especially E167K (glutamic acid to lysine), are also associated with an improved cardiovascular outcome, which is at least in part due to a redistribution of lipids from the periphery to the liver.[94], [98]
Resistance to fibrosis reversal
Only recently, we obtained clear evidence that even compensated cirrhosis is reversible following successful suppression of HBV or eradication of HCV infection.[24], [25] This indicates that once the major liver disease and fibrogenic stimulus is eliminated, progression is not only inhibited, but fibrolysis may prevail, resulting in regression from (compensated) cirrhosis to a normal liver. However, in these and other studies, an estimated 15-25% of patients with compensated cirrhosis due to hepatitis B and C do not regress despite effective antiviral therapies. This percentage appears to be even higher in abstinent patients with alcohol-induced cirrhosis,[99], [100] and data for NASH-related cirrhosis are lacking or inconsistent. The reason for this resistance is unclear, but genetic and epigenetic factors, also affecting hepatic HSC and MF appear to be relevant.101 Thus, two preclinical studies showed that the HSC/MF of mice whose fibrosis was allowed to reverse after induction with carbon tetrachloride CCl4, which occurs spontaneously over several weeks in precirrhotic stages, maintained an epigenetic memory to more quickly resume a profibrogenic phenotype after renewed exposure to CCl4.[102], [103] It is likely that similar epigenetic memories and imprints also operate for other cell types that are involved in fibrosis.
Other cells and factors central to liver fibrogenesis
In early liver disease excess fibrogenesis is matched by removal of excess ECM by proteolytic enzymes (fibrolysis), mainly via the ECM degrading matrix metalloproteinases (MMPs), such as MMP-1, -3, -8, -9, -12, and -13.53 Upon protracted injury, fibrogenesis prevails over fibrolysis, resulting in excess ECM deposition, which is accompanied by a downregulation of MMP secretion and activity, and by an increase of the tissue inhibitors of MMPs (TIMPs), especially TIMP-1, the major physiological inhibitor of most MMPs. However, activation of MMPs at the wrong place and time can also lead to more tissue damage favouring an enhanced fibrogenic response. Kupffer cells/macrophages, proliferating bile ductular epithelia (cholangiocytes, hepatic progenitors), but also endothelia, other inflammatory cells and MFs secrete fibrogenic cytokines and growth factors that can stimulate HSC/MF and perivascular fibroblasts, or further recruit inflammatory cells (Fig. 4).
The role of progenitor cell activation
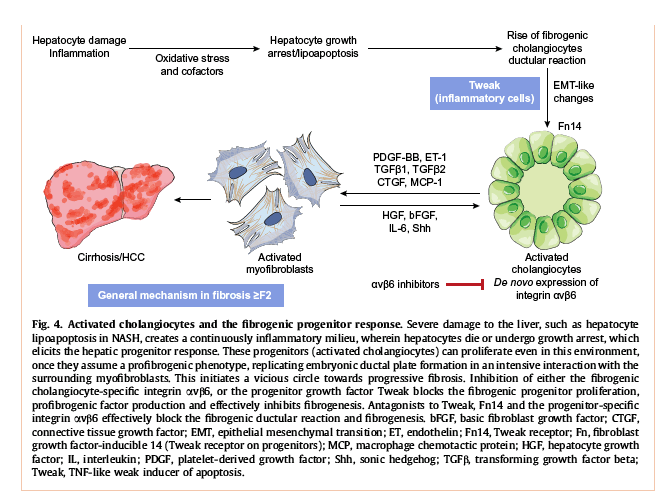
In NASH, the depletion of functional hepatocytes by apoptosis triggers a progenitor (activated cholangiocyte) response aimed at replenishing hepatocytes. However, under the unfavourable condition of chronic inflammation and oxidative stress, hepatocyte differentiation is blocked, and the progenitors develop to a more resistant cholangiocyte-like phenotype, characterised by positivity for cytokeratins CK7 and CK19. These cholangiocytes are strong promoters of fibrogenesis, especially in more advanced stages of NASH, such as stages 2-4. This mechanism that corresponds to the ductular reaction is prominent in lipoapoptotic NASH, when hepatocytes continuously die or enter a state of growth arrest, preventing regeneration by cell division,[104], [105] but is also observed in all other more advanced chronic liver diseases.[106], [107], [108], [109] Here cholangiocyte/progenitor cell proliferation parallels fibrosis progression. It can be seen as an attempt by the injured liver to regenerate the hepatocyte compartment by progenitor production. However, under the unfavourable environment of chronic inflammation, progenitors are forced to differentiate into the more stress resistant fibrogenic cholangiocytes. The activated cholangiocytes/progenitors produce a broad range of central fibrogenic mediators, including transforming growth factor (TGF)β1 and the ductular cell specific TGFβ2,110 platelet-derived growth factor (PDGF)-BB,111 or sonic hedgehog that drive MF/HSC activation.112 On the other hand, the activated HSC/MF produce survival factors for these cholangiocytes, maintaining a program of developmental, fibrogenic ductal plate/portal tract formation. Pharmacological targeting of these fibrogenic progenitors is possible, since they uniquely express the integrin αvβ6 that is an attachment protein for the temporary ECM proteins fibronectin and tenascin-C, and that acts as coactivator of latent TGFβ1, the most potent profibrogenic cytokine. A small molecule inhibitor as well as a blocking antibody to integrin αvβ6 effectively attenuated biliary and non-biliary fibrogenesis via inhibition of fibrogenic progenitor proliferation and TGFβ1 activation.[81], [82], [83], [84] In addition, a factor that promotes unfavourable progenitor activation, TNF-like weak activator (Tweak), could be successfully antagonised with a humanised antibody to promote liver regeneration and inhibit fibrogenesis in cirrhotic mice after partial hepatectomy.113 The role of progenitors/cholangiocytes is summarised (Fig. 5).
The role of macrophages
Apart from targeting the final effector cells of fibrogenesis (HSC/MF), addressing the immune system, particularly macrophages, is an attractive strategy to induce regression of established fibrosis.[53], [61], [63], [50], [51], [54], [66], [114], [115], [116] Among the innate immune cells, liver macrophages, both resident Kupffer cells and freshly recruited monocytes/macrophages play a key role both in fibrogenesis and fibrolysis. After acute injury Kupffer cells can recruit additional innate immune cells, including large numbers of Ly6Chi inflammatory blood monocytes that quickly acquire the macrophage phenotype CD11b+ F4/80+ and display high phagocytotic activity.[117], [118], [119], [120] These cells have the capacity to produce a wide range of cytokines that can act in both a pro- and anti-inflammatory/fibrotic manner, depending on the timing of release, the ECM- and the immune environment.[119], [120] Acute bouts of inflammation induce ECM degradation, paving the way for favourable or unfavourable tissue remodelling, whereas repeat recovery phases in alternation with acute inflammation favour a compensatory immune suppressive but profibrogenic immune response, characterised by high levels of macrophage-derived factors such as interleukin (IL)-13 and TGFβ1, resulting in progressive fibrosis as illustrated (Fig. 2). Conversely, during long-term reversal and in the absence of recurrent bouts of inflammation, macrophages adopt a fibrolytic character characterised by an upregulated MMP-expression, a Ly6Clo F4/80+ phenotype, with an attenuated secretion of TGFβ1.120However, the full functional phenotype of these "pro-resolution" macrophages, which appear to represent a subpopulation of the otherwise profibrogenic M2 macrophages, remains to be better characterised. Therefore, targeting the profibrogenic macrophages, either blocking their recruitment and fibrogenic activation, or pharmacologically switching profibrogenic macrophages into fibrolytic macrophages, by targeting specific receptors or signalling molecules that may induce such a switch, is an attractive therapeutic strategy being actively pursued.[53], [114] The macrophage phenotypes and potential switches relevant to fibrosis reversal vs. fibrosis progression are summarised (Fig. 5).
Antifibrotic therapies
Key point
Therapy that targets the central pathology of a given liver disease is the best strategy for inhibiting fibrogenesis and cirrhosis.
An effective causal therapy that addresses the central pathology of chronic liver disease (the primary hit) is the best strategy to inhibit fibrogenesis and the development of cirrhosis. However, it is not necessarily a good therapy to induce reversal of advanced fibrosis and cirrhosis (fibrosis stage 3-4), which is often characterised by subdued classical inflammation, but an active, continuously reparative fibrotic milieu, as frequently observed in advanced, supposedly non-inflammatory NASH. Therefore, specific antifibrotic therapies are needed for those patients in whom the primary causal factor(s) cannot be eliminated, fibrosis reversal needs to be accelerated (in addition to lifestyle changes or bariatric surgery), or who despite these measures progress or do not regress. Mechanism-based antifibrotic therapies and approaches beyond those that were described in this review have been summarised before.[53], [58], [61], [62], [63] The indication for specific antifibrotic agents and their prime cellular targets are highlighted (Fig. 6).
Conclusion
Advanced fibrosis and cirrhosis have become the major liver-related clinical endpoints in NASH. The natural history of fibrosis progression and regression, and the key underlying genetic and lifestyle factors are already well defined. Clinical studies that address fibrosis will be supported by an improved trial design based on optimised conventional parameters, including improved biopsy readouts, and by a growing armamentarium of surrogate methods and serum markers to assess fibrosis and fibrogenesis. Notably, improved non-invasive markers of fibrogenesis should permit adjustment of drug dose and drug combinations, to generate a maximal antifibrotic potency with minimal side effects. Still, the first clear proof of antifibrotic activity will not be available for another 2-3 years, even for those agents that are currently in phase III clinical studies for NASH. In this vein, a truly personalised approach for treating liver fibrosis would be possible. We anticipate that effective antifibrotic agents for treating patients with advanced liver disease will be available soon.
Financial support
The authors' studies related to this topic were supported by grants from the US National Institutes of Health (NIH), the German Research Foundation (DFG), the German Ministry of Education and Research (BMBF), and an ERC Advanced Grant (FIBROIMAGING) by the European Union to DS the European Union EU to DS.
|
| |
|
|
|
|
|