 |
|
|
| |
Pharmacotherapy for NASH: Current and emerging
|
| |
| |
Download the PDF here
Jnl of Hepatology Jan 2018 - Monica A. Konerman1, Jacob C. Jones2, Stephen A. Harrison3
"Pharmacologic treatment trials in NASH are enrolling patients at a slower pace than treatment trials in other disease states. This is explained by several potential factors including low disease awareness among patients and providers, as well as barriers to enrollment such as complex enrollment criteria and the need for multiple liver biopsies. Until awareness on many levels improves, most patients will go undiagnosed. NAFLD/NASH disease state awareness at the patient, primary care and specialty provider level is very low. Survey studies of knowledge about NAFLD among the general population have consistently shown low levels of awareness. This remains true even in patients with multiple metabolic risk factors who have a high likelihood of having underlying NAFLD.[52], [53] Further complicating this is the fact that many providers perceive NAFLD as a benign entity and subsequently, many patients with a high likelihood of NAFLD do not complete formal evaluation to confirm the diagnosis and are not subsequently referred to specialists who are involved in clinical trial recruitment.[54], [55] Primary care physicians, endocrinologists, gynaecologists, cardiologists and to some extent gastroenterologists lack awareness of NAFLD/NASH progression and its associated risk factors."
Summary
Non-alcoholic fatty liver disease (NAFLD) has become one of the most prominent forms of chronic liver disease worldwide, reflecting the epidemic of global obesity. Those with the progressive variant of NAFLD, non-alcoholic steatohepatitis (NASH), are at significantly increased risk of multisystem morbidity and mortality. However, there are currently no approved pharmacologic therapies for NASH. Given the disease burden, there is an important unmet need for pharmacologic treatment options for this patient population. The underlying pathophysiologic mechanisms that contribute to the development and progression of NAFLD and NASH are complex and reflected by the myriad of therapies, with different targets, currently under investigation. In broad strokes, drug development has focused on modulation of metabolic pathways, inflammatory cascades, and/or mechanisms impacting fibrosis. Although much progress has been made in enhancing our understanding of NAFLD pathogenesis, development of pharmacologic treatments has been hindered by challenges in clinical trial enrollment and complexities in clinical trial design. The compounds in phase IIa have provided promising results in terms of potential benefits on various aspects of histopathology. Agents in later stages of development have shown fairly modest results in terms of reduction of hepatic steatosis, necroinflammation and fibrosis. If longer term safety and efficacy are established among heterogeneous cohorts, these medications may help mitigate potential morbidity and mortality for this burgeoning patient population.
Introduction
Non-alcoholic fatty liver disease (NAFLD) has become one of the most prominent forms of chronic liver disease worldwide, as a direct result of the obesity epidemic. The World Health Organization estimates the prevalence of obesity has more than doubled since 1980 with >600 million people (13%) having a body mass index (BMI) ≥30.1 In this setting, the global prevalence of diabetes among adults has also increased from 4.7% in 1980 to 8.5% as of 2014.2 Within the general population, the overall global prevalence of NAFLD (defined using imaging criteria) is estimated to be 25% (95% CI 22.1-28.6%) though substantial variability was noted across geographic regions (peak prevalence in the Middle East [31.8%] and South America [30.4%], with the lowest rates noted in Africa [13.5%]).3 Prevalence rates of NAFLD among those with metabolic disease are notably higher.
Approximately a third of patients with hypertension, half of patients with dyslipidaemia, up to two-thirds of patients with type II diabetes, and >90% of patients undergoing bariatric surgery had evidence of NAFLD.[4], [5], [6], [7], [8], [9], [10] Additional variations in prevalence rates are attributable to gender (twice as high in men than pre-menopausal women) and ethnicity (higher rates in Hispanic individuals and significantly lower rates in non-Hispanic black individuals), with ethnic variations partly driven by genetic distributions of PNPLA3 among other factors.[11], [12], [13]
Within the broad population of patients with NAFLD, a subset have associated inflammation and hepatocyte injury (with or without accompanying fibrosis) termed non-alcoholic steatohepatitis (NASH). The prevalence of NASH among patients with NAFLD is challenging to assess because of the biopsy-based definition. A review of liver biopsies performed in patients with NAFLD reported NASH prevalence rates ranging from 6.7% to 59% depending on whether the procedure was done in the absence or presence of a specific "clinical indication".3 Focussing on the only prospective prevalence study of NASH to date, the prevalence of NASH among patients presenting for routine colon cancer screening was 12%.14
The public health impact of NAFLD is significant given the worldwide disease burden and the associated morbidity and mortality. Patients with NAFLD are at higher risk of cardiovascular disease, even when accounting for relevant metabolic co-morbidities. In fact, cardiovascular disease represents the leading cause of death for patients with NAFLD.[15], [16] When focussing on liver-related morbidity and mortality, NASH represents the third most common cause of cirrhosis within the United States, but it is predicted to become the leading cause over the next few years.17 NASH cirrhosis is currently the primary aetiology for dual liver-kidney transplantation and is estimated to become the number one indication for liver transplant by 2020.[18], [19], [20] Overall, the distinction between those with NAFLD and those with NASH is clinically relevant given that multiple studies have demonstrated that patients with NASH are at higher risk of adverse liver-related outcomes, with the degree of fibrosis contributing most significantly to this increased risk.[3], [21], [22]
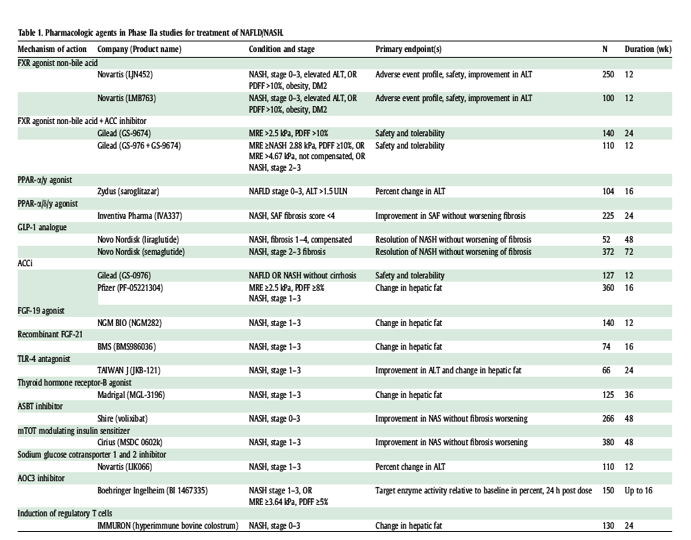
The striking prevalence of NAFLD paired with its profound complications underscores the critical need for safe, effective, and broadly applicable therapy. Presently, there are no medications approved by the Federal Drug Administration (FDA) or European Medicines Agency (EMA) for the treatment of NAFLD or NASH, though as this review will detail, many agents are currently being studied in clinical trials. In current clinical practice, vitamin E is the most commonly used medication, though evidence of efficacy is limited in those with diabetes and cirrhosis and current AASLD and EASL guidelines recommend its use be restricted to patients with NASH in the absence of diabetes and cirrhosis.[23], [24] This recommendation is largely based on data from the PIVENS trial where subjects randomised to 800 IU/day of vitamin E for 96 weeks demonstrated improvements in steatosis (p = 0.005), lobular inflammation (p = 0.02) and ballooning (p = 0.01), as well as resolution of NASH (43% vs. 19% in placebo arm, p = 0.001), but no improvement in fibrosis (p = 0.24).25 There have also been subsequent safety concerns regarding the use of vitamin E, as data suggest a possible increased risk of overall mortality and higher rates of prostate cancer, though this remains controversial.[26], [27], [28]
The insulin sensitiser pioglitazone, a thiazolidinedione, has been well studied and prescribed by some for the treatment of NASH, usually in line with diabetic practice guidelines. Pioglitazone has been extensively evaluated in clinical trials with fairly consistent improvements in various features of NASH and less consistently fibrosis. When evaluated in a meta-analysis, there was an overall improvement in the histopathologic components of NASH (ballooning RR 1.62 and OR 2.11, steatosis RR 2.03 and OR 3.39, and inflammation RR 1.71 and OR 2.58), with improvements in fibrosis (RR 1.38 and OR 1.68).[29], [30], [31] The major downside to the use of pioglitazone is patient and physician acceptance, given its propensity to induce weight gain (average 4.4 kg).25 It should not be used in patients with clinically evident heart failure and may promote post-menopausal bone loss.[32], [33] Given the increasing disease burden and limited efficacy and safety of current treatment options, the development of additional pharmacologic therapies to treat NASH is critical. This review will highlight the process of therapeutic clinical trial design, challenges in clinical trial recruitment, metabolic and antifibrotic agents under investigation, and an overview of the future landscape of pharmacologic design once initial agents are approved.
Endpoints in clinical trials on NAFLD and NASH
Key point
Determining optimal yet feasible endpoints for clinical trials evaluating pharmacologic agents for NAFLD and NASH is complicated by slow progression of clinically significant outcomes.
Determining optimal yet feasible endpoints for clinical trials evaluating pharmacologic agents for NAFLD and NASH is complex, because of the chronic nature of the disease, with typically slow progression to clinically significant outcomes.34 Subsequently, relevant and acceptable surrogate endpoints need to demonstrate efficacy before the onset of long-term complications. In addition, the interconnected and dynamic nature of metabolic, inflammatory and fibrotic aspects of the disease, in response to an intervention, add to the difficulty in identifying clear and precise endpoints. Although the presence of NASH has been clearly linked with fibrosis development, an individual treatment can have different impacts on these two endpoints (i.e. treatment with drug X can improve NASH but worsen fibrosis and vice versa). Moreover, during disease progression fibrosis may worsen, while features of steatohepatitis resolve or "burnout". As a result, NASH and fibrosis need to be evaluated independently to ensure a beneficial impact on one parameter does not simultaneously result in a negative impact on another endpoint of interest, particularly given that individual treatments tend to focus on one primary mechanism of action (i.e. metabolic or NASH disease modifier vs. antifibrotic). Moreover, many of the outcomes of interest involve histological parameters, which pose a unique set of barriers and limitations. This includes inherent limitations of liver biopsy such as interobserver reliability and sampling error. Early phase trials are investigating whether relevant or predictive information can be derived from shorter-term endpoints to inform future trials. The challenge remains with long-term registration trials, which are required to demonstrate that clinically beneficial outcomes are in line with FDA and EMA requirements. This has led to the use of surrogate endpoints for accelerated approval in the US and conditional approval in Europe.
Histological endpoints
Histological endpoints to assess response to a therapeutic intervention can be broadly divided into those meeting a numerical reduction in one of two accepted scoring systems: NAFLD Activity Score (NAS) or Steatosis Activity and Fibrosis (SAF) score or the resolution of NASH as determined by a qualified hepatopathologist.[35], [36] When change in the NAS is used as a primary outcome, it is recommended that ≥2 point improvement in total score be achieved with contribution from multiple parameters of the NAS, alongside no worsening of fibrosis.37 For trials evaluating resolution of NASH, this is defined as a complete resolution of hepatocyte ballooning, with inflammation scores of 0 or 1, in addition to no worsening of fibrosis, though there is no specific requirement regarding change in steatosis. Clinical trials with a focus on improvement in fibrosis must similarly require no worsening in NASH.
Biomarkers as potential surrogates for histology
Because of the limitations and concerns regarding histological endpoints, there is mounting interest in identifying study endpoints that utilise non-invasive technologies for assessment of fat, inflammatory and fibrotic changes in response to a therapeutic intervention (Fig. 1). Promising diagnostic algorithms are being developed to identify patients at risk, with a certain level of NASH severity and fibrosis.38 Imaging assessments are gaining momentum as a surrogate for liver histopathology, although it is premature to comment on when and if this technology will replace histopathology. Non-invasive methods to identify hepatic steatosis include controlled attenuation parameter (CAP) assessment as part of vibration controlled transient elastography (Fibroscan™), and MRI based imaging in the form of proton density fat fraction (PDFF) and multiparametric MRI (LiverMultiscan™). In this scenario, Fibroscan™ CAP may be easier to obtain, but is not as accurate at quantifying grades of steatosis as MRI.39Furthermore, the accuracy with which CAP reflects histologic steatosis improvement over time has not been demonstrated. These novel non-invasive biomarkers are being used in clinical trials, and will need further data and regulatory qualification before they can replace a liver biopsy, both in clinical trials and subsequently in clinical practice.
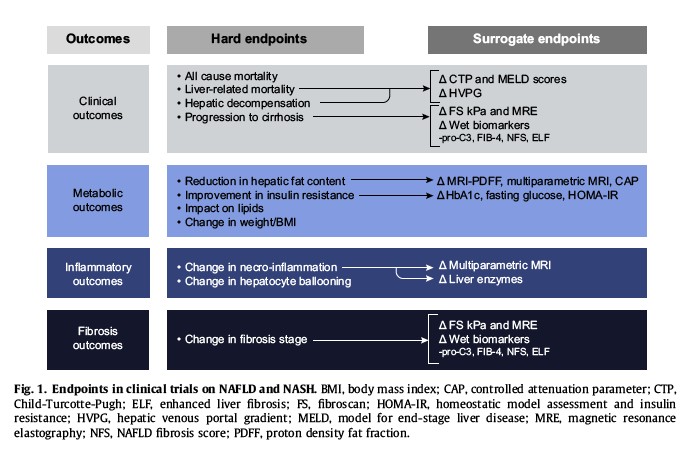
At present, there are a limited number of non-invasive tools to accurately assess changes in liver inflammation/ballooning. Measurements of liver-associated enzymes, particularly alanine aminotransferase (ALT) and aspartate aminotransferase (AST) have historically been utilised and should be obtained. Markers of apoptosis, such as CK-18, have been studied, but may not accurately reflect change in histology over time. Recently, multiparametric MRI, such as LiverMultiScan™, has gained attention for assessment of necroinflammation and shows promise in this area, although more confirmatory data is needed.40
Biomarker assessment of fibrosis is still limited as we are unable to provide a precise assessment of the directionality of fibrosis over time, because of the dynamic balance between fibrogenesis and fibrinolysis. When analysing agents that are focussed on modulating fibrotic pathways, imaging based biomarkers such as transient elastography, multiparametric MRI or magnetic resonance elastography (MRE) can be applied.[39], [41], [42] In this setting, it appears that MRE may be the most accurate method for fibrosis assessment. Further study is needed to determine the accuracy of each of these methods for assessing fibrosis change with therapy and the variation with time (coefficient of variation). Among the clinical and serological or so called "wet biomarkers" and other non-invasive tools for fibrosis assessment, pro-C3, Fibrosis-4, NAFLD fibrosis score and the enhanced liver fibrosis (ELF) score (an algorithm of hyaluronic acid, amino-terminal propeptide-of-type-III-collagen, tissue-inhibitor of matrix metalloproteinase-1) appear to have promise.[43], [44], [45], [46]
Clinical endpoints
As discussed earlier, therapies ultimately need to demonstrate efficacy on long-term clinical outcomes. Prior studies have estimated that from a clinical outcomes perspective, approximately one-third of patients have stable disease, one-third have progressive disease and one-third have improved disease stage over 14 and 7 years, for patients with NAFLD and NASH, respectively.47 The indolent and dynamic bidirectional nature of disease progression poses challenges in the assessment of these outcome types among individuals with early stage disease. The most appropriate endpoints vary depending on the stage of disease being studied. In the setting of cirrhosis, relevant endpoints include changes in degree of portal hypertension or the development of hepatic decompensation. In clinical trials of cirrhosis, all-cause and liver-related mortality are also important and tenable outcomes.37 Changes in portal hypertension, which is tightly linked to clinical outcomes, can be assessed directly through measurement of the hepatic venous pressure gradient (HVPG) or determined by the impact of the intervention on liver-related events. To evaluate changes in hepatic synthetic function and the development of hepatic decompensation, serial monitoring of Child-Turcotte-Pugh (CTP) score, model for end-stage liver disease (MELD) score, and new onset or worsening of liver-related events such as ascites, variceal bleeding, hepatic encephalopathy and HCC are needed. Other non-invasive serial monitoring tests to assess changes in liver function are being investigated, including the methacetin breath test and HepQuant.[48], [49]
Safety
Drug induced liver injury and other toxicity signals will make an otherwise viable therapeutic agent untenable and this needs to be determined as early in development as possible. Therefore, early phase clinical trials must carefully assess safety and tolerability. Since NASH is a largely asymptomatic disease, a drug with a burdensome side effect profile may not be acceptable. Furthermore, a viable potential treatment cannot adversely impact cardiovascular risk, as this is the most common cause of death in patients with NASH.
Determining the appropriate clinical trial duration
NASH is a chronic disease like diabetes and it is likely that any intervention will need to be potentially life-long, though this may depend on the drug's mechanism of action. Similarly, careful consideration of the drug's mechanism of action and target endpoint in the context of the phase of development will determine the appropriate trial length. As a result, determining the appropriate duration of clinical trial length is a balance between having sufficient time to reach the efficacy endpoint and the need to continually assess the risk of chronic exposure over longer periods. In general terms, phase II trials are of shorter duration since they are meant to provide an efficacy signal before proceeding to a pivotal trial. Subsequently, phase III trials typically test more definitive endpoints for a longer duration, often extending multiple years. Post market approval, surveillance of patients on long-term therapy will be critical to ensure long-term safety, tolerability and sustained effect. There are many unknowns regarding the natural history of liver disease progression in NASH, thus determining the time course for overall outcomes assessment (including liver-related outcomes, adverse event profiles and safety) may be imprecise until more robust data are available.
Developmental pathway: phase IIA, IIB, and III
Key point
The design of each phase of a clinical trial for a potential therapeutic agent is strongly influenced by the drug's mechanism of action.
In NASH, the developmental pathway for therapeutic agents involves navigating multiple complexities that include the non-linear, heterogeneous nature of disease progression, bi-directionality of disease state and the limitations in identifying clinically relevant shorter-term outcomes, as previously outlined. The design of each phase of a clinical trial for a potential therapeutic agent is strongly influenced by the drug's mechanism of action (Fig. 2). In addition, characteristics of the patient population under study heavily influence trial design. In this review, we have only focussed on adult studies as paediatric drug development pathways differ significantly. Phase IIa trials focus on proof of concept (POC) and short-term safety. As a result, phase IIa trials have truncated time courses of evaluation. This creates challenges for the selection of relevant endpoints given that pharmacologic agents for the treatment of NASH and especially fibrosis have a low likelihood of inducing clinically significant improvements over this short time frame. In addition, paired liver biopsies performed at short intervals can raise ethical concerns given the potential risk to the patient. In the absence of histology, phase IIa studies should employ early surrogate endpoints that are aimed at confirming target engagement of the experimental compound, in addition to safety. It is important to understand that these non-invasive markers are currently unapproved surrogates for histology and simply allow for initial POC testing for target engagement.
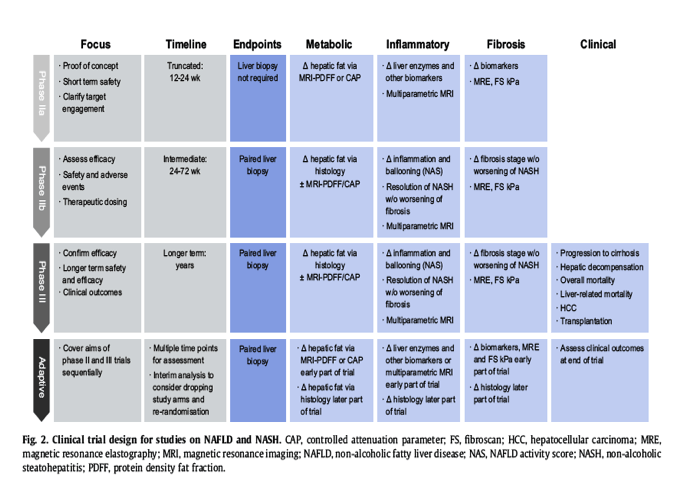
Assuming that phase IIa trials provide encouraging results, subsequent phase IIb and phase III studies should focus on confirming therapeutic dose, verifying clinical efficacy via histology and assessing long-term clinical outcomes of NASH patients. Large clinical trials with paired biopsy designs provide an invaluable opportunity to test non-invasive disease surrogates that may someday supplant histology or provide an early efficacy or futility signal. Surrogate non-invasive endpoints could be assessed along the way with paired histology as well. After initial approval, subsequent phase IIIb clinical outcome studies continue to evaluate longer term safety and efficacy. Adaptive trial designs may be a feasible option to overcome some of the existing complexities in clinical trial design for NASH. Adaptive designs allow for planned modifications in one or more aspects of the study design based on response in earlier phases. This approach can potentially limit the total number of patients needed to enroll over the entire process of individual drug development, by allowing patients to transition through multiple phases of the study (Fig. 2).[37], [50], [51] Based on results of interim analyses, preplanned adaptations such as discontinuing specific study arms, randomising additional patients, or stopping the study entirely can be implemented.
Current challenges in clinical trial recruitment
Pharmacologic treatment trials in NASH are enrolling patients at a slower pace than treatment trials in other disease states. This is explained by several potential factors including low disease awareness among patients and providers, as well as barriers to enrollment such as complex enrollment criteria and the need for multiple liver biopsies. Until awareness on many levels improves, most patients will go undiagnosed. NAFLD/NASH disease state awareness at the patient, primary care and specialty provider level is very low. Survey studies of knowledge about NAFLD among the general population have consistently shown low levels of awareness. This remains true even in patients with multiple metabolic risk factors who have a high likelihood of having underlying NAFLD.[52], [53] Further complicating this is the fact that many providers perceive NAFLD as a benign entity and subsequently, many patients with a high likelihood of NAFLD do not complete formal evaluation to confirm the diagnosis and are not subsequently referred to specialists who are involved in clinical trial recruitment.[54], [55] Primary care physicians, endocrinologists, gynaecologists, cardiologists and to some extent gastroenterologists lack awareness of NAFLD/NASH progression and its associated risk factors. Once patients have the opportunity to enroll in a clinical trial, multiple barriers complicate patient acceptance and subsequent enrollment. Because of a lack of reliable surrogates and reliance on histology, many studies require multiple liver biopsies, particularly in the more advanced phase trials. The sheer volume of clinical trials in this disease space has also contributed to slow enrollment. Finally, due to the complex and heterogeneous nature of the disease, the inclusion and exclusion criteria for enrollment in NAFLD/NASH clinical trials have made it challenging to enroll. Many trials have restrictions on age, BMI, ALT, AST, haemoglobin A1c and bilirubin elevations. Restrictions on patients with a history of cancer or bariatric surgery, as well as on those taking concomitant medications have further hampered enrollment.
Once patients with established NAFLD are identified, there are additional challenges in identifying the subset of patients with NASH and fibrosis that are eligible for enrollment in current treatment trials. Liver biopsy is infrequently performed in clinical practice and many patients without a pre-existing biopsy are hesitant to have one performed. One potential option for streamlining enrollment and limiting screening failures is to apply a risk stratification approach, that uses data gleaned from studies investigating risk factors for the presence of NASH with fibrosis in the larger pool of patients with NAFLD (Fig. 3).[56], [57], [58] While the ideal algorithm to identify patients with NASH remains to be found, this approach provides some general guidance that may help to stratify patients into high, intermediate and low likelihood of meeting enrollment criteria.
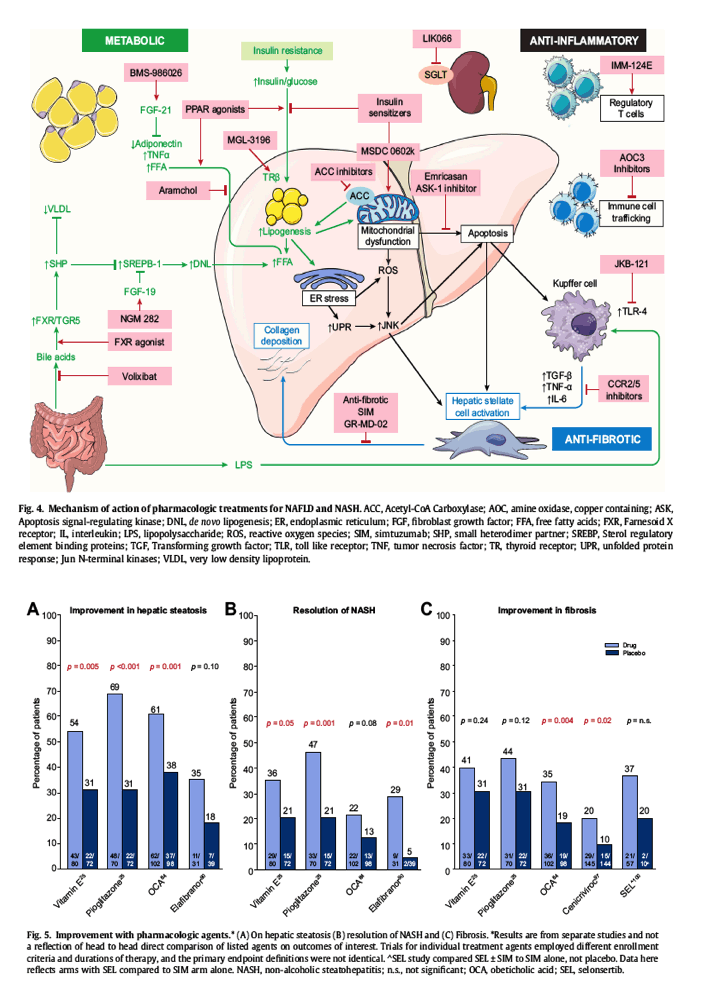
NASH is a heterogeneous disease with a correspondingly complex pathophysiology, which includes redundant pathways that may not be uniform across patients. This heterogeneity and complexity has made finding a single agent to effectively treat most patients a challenge. However, its complex pathophysiology permits the development of a wide array of potentially viable therapeutic targets (Fig. 4). A concise summary of the efficacy of pharmacologic agents in later stages of development is displayed (Fig. 5), with more detailed description of the individual agents and clinical studies provided later.
Phase IIB and IIA pharmacologic agents
Key Point
Only a small proportion of pharmacologic treatments evaluated in phase II progress to phase III and even fewer progress to market.
There are currently multiple agents in phase IIa and IIb clinical trials. However, only a small proportion of pharmacologic treatments evaluated in phase II progress to phase III and even fewer progress to market. The following section attempts to summarise the many compounds in development with the caveat that this is a rapidly evolving landscape and this represents a snapshot in time. Available published data are summarised below and in cases where preliminary human data are not yet in published form, these studies are summarised (Table 1). It is important to note the significant differences in inclusion and exclusion criteria that, as mentioned, are largely driven by the mechanism of action employed by each treatment agent being investigated.
ACC, acetyl-coA carboxylase; ALT, alanine aminotransferase; AOC3, amine oxidase copper containing-3; ASBT, apical sodium dependent bile acid transporter; DM2, type 2 diabetes; FGF, fibroblast growth factor; FXR, farnesoid X receptor; GLP-1, glucagon-like peptide-1; MRE, magnetic resonance elastography; mTOT, mitochondrial target of thiazolidinediones; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; PDFF, proton density fat fraction; PPAR, peroxisome proliferator-activated receptor; TLR, toll-like receptor.
Phase IIA
There are many pharmacologic agents in phase IIa clinical trials, but only a few have presented or published preliminary results. NGM282 is an engineered variant of human fibroblast growth factor (FGF)-19 that works via FGFR4 and FGFR1c to modulate bile acid synthesis and affect multiple metabolic pathways. A recent phase IIa, randomised, placebo-controlled trial enrolled 82 patients with biopsy confirmed NASH (NAS ≥4 with one point in each component, with fibrosis stage 1/2/3) with a minimum of 8% absolute liver fat content by MRI-PDFF and abnormal ALT (≥19 IU/L females, ≥30 IU/L in males). Subjects were randomised to 3 mg of NGM 282 (n = 27), 6 mg of NGM 282 (n = 28), or placebo for 12 weeks. The primary endpoint was change in hepatic steatosis (decrease in absolute liver fat ≥5%).59 There was a significantly higher proportion of patients in the treatment arm who achieved the primary outcome than in the placebo arm (79% vs. 7%). Of those in the treatment arm, 34% achieved normal (<5%) liver fat content (compared to none in placebo). ALT also normalised in 35% and 37% of subjects in the 3 mg and 6 mg arms compared to an average 2% reduction in the placebo arm (p <0.001), reductions that correlated with decreases in liver fat content.
BMS-986026 is a recombinant pegylated FGF21 analogue that has also been identified as having an effective role in metabolic regulation. This FGF21 analogue has beneficial effects on metabolism via increased adiponectin expression, improved insulin sensitivity and decreased lipogenesis. A phase IIa, double-blind, placebo-controlled study, enrolled patients with biopsy proven NASH with fibrosis stage 1-3, BMI ≥25, and hepatic fat fraction ≥10% on MRI-PDFF. Patients were randomised to 10 mg of BMS-986026 daily (n = 25), 20 mg every week (n = 23), or placebo (n = 26) for 16 weeks and assessed for decreases in hepatic fat fraction.60 Patients in the treatment arms had significantly reduced absolute hepatic fat fraction (-6.8 in the 10 mg daily arm, -5.2 in the 20 mg weekly arm, vs. -1.3 in the placebo arm; p = 0.008) and improvements in serum ALT. There were also some improvements in surrogate markers of fibrosis measured by MRE (36% and 33% of patients in the 10 mg daily and 20 mg weekly arms, respectively, had ≥15% reduction in liver stiffness compared to 7% in the control arm) and pro-C3, as measured by adjusted percent improvement from baseline (30%, 21% and 1.9% in the 10 mg daily, 20 mg weekly, and placebo arm, respectively). Improvements in metabolic parameters (adiponectin and lipids) were also seen.
Glucagon-like peptide-1 (GLP-1), a member of the glucagon peptide family, promotes several beneficial traits, including reduced hepatic steatosis and insulin resistance, as well as increased weight loss.61GLP-1 analogues such as liraglutide and semaglutide are currently being utilised for the treatment of type 2 diabetes, and are being evaluated in NASH. The LEAN trial, a phase II, randomised, placebo-controlled trial evaluated subcutaneous injections of liraglutide (1.8 mg daily) (n = 23) compared to placebo (n = 22) for 48 weeks in overweight subjects with NASH. The primary endpoint was resolution of NASH without worsening of fibrosis. In the treatment arm, resolution of NASH was observed (p = 0.019) and ballooning improved (p = 0.05), but fibrosis worsened (p = 0.04).62 Based on these results, semaglutide, but not liraglutide, is now being assessed in a larger phase IIa multi-centre study.
Acetyl-CoA carboxylase (ACC) inhibitors are of interest given that ACC activation promotes lipid storage via stimulation of lipogenesis and inhibition of lipid oxidation.63 Mouse models of ACC inhibitors have shown that these agents can reduce liver triglycerides, but were also noted to increase plasma triglycerides.64 Two ACC inhibitors are currently in phase IIa trials (GS-0976 and PF-05221304).65 Preliminary data from a 12-week clinical phase IIa trial of GS-0976 is available. In this trial, 10 patients with NASH were given 20 mg of GS-0976 daily. There were statistically significant improvements in liver fat content and non-invasive markers of fibrosis. Specifically, there was a median relative decrease in liver fat content measured by MRI-PDFF (15.7% to 9%, p = 0.006), and a reduction in median liver stiffness from 3.4 to 3.1 kPa (p = 0.049) assessed by MRE.66 A separate phase II, randomised, placebo-controlled trial evaluating GS-0976 in 127 subjects with NASH is underway to further assess safety and efficacy.67
Hyperimmune bovine colostrum has been demonstrated to regulate the body's immune response via induction of regulatory T cells, which in turn impact inflammation and insulin resistance.68 Data from an initial trial of 10 patients with NASH and insulin resistance treated with IMM-124E for 30 days resulted in improvements in insulin resistance and lipid profiles and increased levels of T regulatory cells.69 This is being further investigated in a randomised, placebo-controlled, phase II trial in 130 patients with NASH.70
Phase IIB: Aramchol
Aramchol is a novel synthetic fatty acid/bile acid conjugate that impacts lipogenesis. Aramchol upregulates the ABCA1 reverse cholesterol transporter and functions as a partial inhibitor of stearoyl-CoA desaturase 1 (SCD1), a rate-limiting enzyme in the biosynthesis of monounsaturated fatty acids in the liver. In a phase II, randomised, placebo-controlled trial, six patients with biopsy proven NASH and 60 patients with NAFLD were evaluated. The primary endpoint was a change in hepatic steatosis by nuclear magnetic resonance spectroscopy (NMRS). In this trial, patients were randomised 1:1:1 to aramchol 300 mg/day, 100 mg/day, or placebo for 12 weeks.71 Evaluation at month three showed a relative improvement in liver fat content of 12.57% ± 22.14 compared to 6.39% ± 36.27 in the placebo group (p = 0.02). These findings are being further evaluated in a phase IIb, randomised, placebo-controlled trial in overweight/obese adults with biopsy confirmed NASH, and type II diabetes/prediabetes. In this trial, patients were randomised 2:2:1 to aramchol 400 mg/day, 600 mg/day, or placebo for 52 weeks with an additional 13-week follow-up period. The primary outcome was percentage change in liver triglyceride content measured by NMRS. Secondary outcomes of interest included: the proportion of patients with a NAS improvement, without worsening of NASH; the proportion with a NAS improvement ≥2, without worsening of fibrosis; the proportion with SAF score improvement ≥2, without worsening of fibrosis; the proportion with NASH resolution (ballooning 0, inflammation 0 or 1), without worsening of fibrosis and change in ALT.72
Phase IIB: Emricasan
Emricasan is a pan-caspase inhibitor that has been shown to decrease portal hypertension by blocking apoptotic and inflammatory caspase activation involved in hepatocyte cell death.73 In a phase IIa study of patients with a baseline HVPG of 12 mmHg, patients treated with emricasan over 28 days had a 17.2% decrease in HVPG and a significant decrease in AST and ALT levels.74 These findings are being further explored in multiple phase IIb studies. ENCORE-NF is a multicentre, randomised, placebo-controlled trial of patients with biopsy proven NASH and fibrosis, but not cirrhosis, that has completed enrollment. In this study, patients were randomised to 5 mg vs. 50 mg vs. placebo for 72 weeks, with a primary endpoint of improvement in fibrosis ≥1 stage without worsening of steatohepatitis.75 Two subsequent phase IIb trials are also underway in NASH patients with either cirrhosis and severe portal hypertension (HVPG ≥12 mmHg) or decompensated cirrhosis. These studies will look for improvement in HVPG over 24 weeks or improving event-free survival relative to placebo, respectively.[76], [77]
Phase IIB: GR-MD-02
Galectin-3 is a protein expressed in immune cells that has been shown to play a key role in fibrogenesis. GR-MD-02 is a carbohydrate that functions as a galectin-3 protein inhibitor. Animal studies using galectin-3 protein inhibitors have demonstrated a decrease in fibrosis which has subsequently been replicated in phase I human studies.78 An initial phase IIa trial of GR-MD-02 given over 16 weeks assessed change in fibrosis by MRI in 30 patients with biopsy proven NASH and stage 3 fibrosis.79 Although this study did not achieve its primary endpoint, this agent is undergoing subsequent evaluation in a longer term (one year), randomised, placebo-controlled, phase IIb trial in patients with compensated NASH cirrhosis and portal hypertension (HVPG ≥6 mmHg). Patients receive GR-MD-02 IV every two weeks or placebo for 52 weeks.80 Enrollment has completed and results are expected towards the end of 2017.
Pharmacologic agents currently in phase III of development
Obeticholic acid
Obeticholic acid (OCA) is a semi-synthetic derivative of the primary human bile acid chenodeoxycholic acid (CDCA) and functions as an agonist of the farnesoid X receptor (FXR). OCA downregulates hepatic glucose and lipid metabolism by downregulating SREPB-1c and reducing endogenous bile acid production, via upregulation of FGF19 which then inhibits CYP7A1, the rate-limiting enzyme for the conversion of cholesterol to bile acids.81In addition to these primary metabolic effects, OCA may also decrease portal pressure by increasing iNOS and displaying a wider range of anti-inflammatory and antifibrotic activity.[82], [83] The FXR Ligand Obeticholic Acid in NASH Treatment (FLINT) trial was a randomised, placebo-controlled, phase IIb trial to evaluate the safety and efficacy of OCA in NASH. Patients enrolled in the trial had biopsy proven NASH/borderline NASH (NAS ≥4) without cirrhosis (n = 283). Patients were randomised 1:1 to either OCA 25 mg daily or placebo for 72 weeks. In this study, 45% of patients in the treatment arm had at least a 2-point improvement in the NAS without worsening of fibrosis, compared to only 21% in the placebo arm (p = 0.0002). Of note, the proportion of patients who achieved resolution of NASH (defined as either not NAFLD or NAFLD but not NASH) was not statistically different (22% for OCA vs. 13% for placebo, p = 0.08). Interestingly, OCA did have a beneficial impact on fibrosis (35% of patients in the treatment arm vs. 19% in the placebo arm had improvements in fibrosis, p = 0.004), though this did not translate into significant resolution of advanced fibrosis in those receiving OCA.84 Two adverse events occurred more commonly in those receiving OCA compared to placebo; pruritus (23% vs. 6%, p <0.0001) and an unfavourable alteration in lipid profile, with an increase in low-density lipoprotein (LDL) and decrease in high-density lipoprotein (HDL). These potential metabolic effects will need to be further characterised given the risk profile of the patient population of interest. Currently OCA is being evaluated in a large phase III, randomised, placebo-controlled trial termed REGENERATE. The aim of this trial is to evaluate longer term safety and efficacy of OCA for improvement of NASH and fibrosis. This long-term trial has target enrollment of around 2,000 patients with biopsy confirmed NASH and fibrosis stage 1-3 (with the exception of stage 1c). Individuals are randomised 1:1:1 to OCA 25 mg/day vs. 10 mg/day vs.placebo. For the purpose of FDA subpart H approval, at 18 months, a histopathologic interim analysis will be performed on the first 750 enrolled patients with fibrosis stage 2-3, to assess for fibrosis improvement ≥1 point without NASH worsening, or NASH resolution without worsening of fibrosis. Subsequent analysis via liver biopsy will occur at 48 months, with final assessment based on an estimated number of outcome events at year six. Outcome events of interest include time to: histological progression to cirrhosis, uncontrolled ascites, HCC, liver transplantation/transplant eligibility, and hospitalisation for hepatic decompensation or death.85
Key Point
Currently OCA is being evaluated in a large phase III, randomised, placebo-controlled trial
Elafibranor
Elafibranor is another agent currently in phase III for the treatment of NASH. Elafibranor is a dual peroxisome proliferator-activated receptor (PPAR)-α/σ agonist and as such has effects on the regulation of metabolic homeostasis, inflammation, cellular growth, and differentiation.86 PPAR-α is a regulator of fatty acid transport and a major inducer of β-oxidation of fatty acids. In animal models, PPAR-α expression significantly reduces cholesterol, triglycerides, and free fatty acids while increasing HDL cholesterol levels and decreasing overall hepatic inflammation, though ostensibly peroxisomes are less abundant in humans.[86], [87] PPAR-σ reduces fatty acid uptake, and alters glucose homeostasis, energy metabolism and hepatic inflammation in the liver. PPAR-σ is also highly expressed in adipose tissue where it controls adipocyte differentiation and plays a crucial role in increasing insulin sensitivity and promoting fatty acid uptake into adipocytes.[88], [89] The phase IIb trial of elafibranor, GOLDEN-505, was an international, randomised, placebo-controlled trial of 276 patients with biopsy proven NASH in the absence of cirrhosis, with a primary outcome of resolution of NASH defined by disappearance of steatosis, ballooning or lobular inflammation. Patients were stratified by presence of diabetes and randomised to elafibranor 80 mg daily vs. 120 mg daily vs. placebo for 52 weeks.90 The difference in primary outcome was not statistically significant across treatment arms (23%, 21% and 17% respectively, p = 0.28). However, when re-evaluated using the more stringent definition of NASH resolution now recommended by regulatory authorities (disappearance of ballooning, none or mild persistence of lobular inflammation) there was a significant difference across treatment arms for the 120 mg dose (13% vs. 19% vs. 12%, p = 0.045). Changes in ballooning and lobular inflammation were also noted to correlate with changes in fibrosis stage.91 Elafibranor significantly decreased total cholesterol, LDL-cholesterol, triglycerides, and increased HDL-cholesterol, and significantly improved glucose homeostasis (significant reductions in HbA1c and free fatty acids in diabetic patients). Elafibranor was well tolerated, but did produce a mild, reversible increase in serum creatinine. These findings are being investigated further in a randomised, placebo-controlled, phase III trial of patients with biopsy proven NASH (NAS ≥4) and fibrosis stage 1-3, RESOLVE-IT. A targeted 2,000 patients will be randomised 2:1 to elafibranor 120 mg daily vs. placebo. Interim analysis, at week 72, of 1,000 patients with fibrosis stage 2-3 will assess for the primary endpoint of NASH resolution without worsening of fibrosis, for the purposes of FDA subpart H approval. The study is estimated to last until accrual of a pre-specified number of events of interest: all-cause mortality, cirrhosis development, and liver-related clinical outcomes (estimated duration four years of treatment).92
Cenicriviroc
Cenicriviroc (CVC) is a dual CCR2/CCR5 chemokine receptor antagonist that has been shown to play key roles in hepatic inflammation and fibrosis.[93], [94] In the setting of hepatocyte injury, kupffer cells secrete C-C chemokine ligand type 2 (CCL2) triggering CCR2 receptors to initiate an inflammatory response. This inflammatory process involves macrophage secretion of pro-inflammatory cytokines, platelet-derived growth factor, and interlukin-1β. This also activates pro-inflammatory mediators in adipose tissue which together ultimately activate hepatic stellate cells.[94], [95], [96]CVC was evaluated in a phase IIb, randomised, placebo-controlled, multinational study of 289 patients with biopsy proven NASH (NAS ≥4, with fibrosis stage 1-3) and diabetes or metabolic syndrome (CENTAUR). Patients were randomised to CVC 150 mg daily for two years, vs. placebo for one year followed by 150 mg daily in year two, vs. placebo for two years. The primary endpoint was a 2-point improvement in the NAS without worsening of fibrosis. In analysis of one-year data, rates of achievement of the primary endpoint were not statistically different (16% CVC vs. 19% placebo, p = 0.52). Rates of resolution of steatohepatitis were also similar (8% CVC and 6% placebo, p = 0.49), although twice as many patients in the CVC arm had improvement in fibrosis by ≥1 stage without worsening in steatohepatitis (20% vs. 10%, p = 0.02).97 This impact on fibrosis is being further investigated in a current phase III clinical trial of patients with NASH and fibrosis stage 2-3. In this two arm trial, patients will be randomised to CVC 150 mg vs. placebo for 12 months with a primary outcome of improvement in fibrosis by ≥1 stage and no worsening of steatohepatitis.98
Selonsertib
Apoptosis signal-regulating kinase 1 (ASK1) is a large contributor to the pathogenesis of oxidative stress-related cell death, fibrosis and inflammation. ASK-1 is activated by reactive oxygen species, tumour necrosis factor-α, endoplasmic reticulum stress, and lipopolysaccharide. Once activated, ASK1 in turn activates mitogen-activated protein kinase (MAPK) that consequently activates c-Jun N-terminal Kinase (JNK).99 An apoptosis signal-regulating kinase 1 (ASK1) inhibitor [GS-4997- selonsertib (SEL)] is being studied in patients with NASH. The phase IIb randomised trial of 67 patients with biopsy proven NASH, with NAS ≥5 and fibrosis stage 2-3, evaluated change in fibrosis stage as a primary endpoint. Patients were randomised 2:2:1:1:1 (stratified by diabetes) to SEL 6 mg daily ± simtuzumab (SIM) 125 mg subcutaneously (sq) weekly, vs. SEL 18 mg daily ± SIM 125 mg sq weekly, vs. SIM 125 mg sq weekly for a total of 24 weeks. Based on efficacy results from prior studies, for the purpose of this trial SIM was considered equivalent to "placebo". Overall 43% of subjects in the SEL 18 mg ± SIM arm had improvement in fibrosis compared to 30% in the SEL 9 mg ± SIM arm, vs. 20% in the SIM alone arm.100 There are currently two phase III trials of SEL, exploring its effectiveness in patients with bridging fibrosis and compensated NASH cirrhosis. In both trials the primary endpoint is improvement in fibrosis by ≥1 stage without worsening of NASH.[101], [102]
Combination therapies
Key Point
It is likely that combining therapies that engage different targets may provide a synergistic histopathologic benefit.
Presently the majority of NASH clinical trials are being conducted with single agent therapies. However, based on the complex pathophysiology and presumed multiple redundant pathways, it is likely that combining therapies that engage different targets may provide a synergistic histopathologic benefit. Presently the combination of a non-bile acid FXR agonist (GS-9674) + ACC inhibitor (GS-0976) is being investigated in an early phase II clinical trial.103 In an animal model of NASH, treatment with this combination therapy demonstrated a greater reduction in hepatic steatosis and genes associated with fibrosis than treatment with either agent alone.104 Future clinical trials will likely use combinations of agents that have already produced positive results individually. Logistically, these combination therapies are most easily studied among compounds manufactured by the same pharmaceutical company, though combinations of promising agents across companies may also be considered to maximise the benefit of individual therapies (i.e. combinations of an agent with positive metabolic effects and an agent with significant antifibrotic effects).
Future of clinical trials once therapies are approved
With multiple agents currently in phase III clinical trials, it is important to consider how the eventual approval of a pharmacologic agent for NASH will impact ongoing and future clinical trials in this arena. Potential implications include the incorporation of the regulatory approved agent as a control arm in future clinical trials. Alternatives include adjustment of primary endpoints to demonstrate improvement in metabolic and fibrotic outcomes above what is achieved by the approved therapies.
Conclusions
There are presently no approved pharmacologic agents for the treatment of NAFLD and NASH, but numerous agents are currently being investigated in phase II and III clinical trials. Data from these studies has shown promising improvements in steatosis, inflammation and fibrosis. Because of the longitudinal and heterogeneous nature of disease progression in NAFLD/NASH, investigation into the long-term efficacy and safety of these agents will be critical, particularly given concerns raised for potential adverse metabolic effects of some of these agents. Addressing important barriers to clinical trial enrollment and optimising clinical trial design in NAFLD/NASH will also be key for approval of therapeutic agents for this disease. Incorporation of non-invasive parameters to assess steatosis, inflammation and fibrosis in addition to considering adaptive trial designs are potential approaches.
|
| |
|
|
|
|
|