 |
|
|
| |
Fatty Liver, NASH - Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis
|
| |
| |
Download the PDF here
"The economic burden of nonalcoholic fatty liver disease in the United States is already enormous, with a current estimate of more than $100 billion in annual direct medical costs, much of which is attributable to nonalcoholic steatohepatitis and its sequelae.....the heavy societal burden imposed by liver damage related to nonalcoholic steatohepatitis justifies public health measures aimed at eradicating nonalcoholic steatohepatitis and other metabolic syndrome-related diseases by encouraging healthful eating, facilitating exercise and fitness, limiting shift work, and minimizing exposure to environmental toxins.....A number of inherited and environmental factors increase the risk of nonalcoholic steatohepatitis and influence its progression. The pathogenic mechanisms are being unraveled; metabolic stress, inflammation, and fibrosis have been identified as key processes. Pharmacologic agents that target these mechanisms are being investigated in clinical trials."
NATAP Fatty Liver http://www.natap.org/liver.htm
Current and Future Therapeutic Approaches to NAFLD/NASH - (08/05/17)
EASL: Emerging Treatments for ASH & NASH - Rohit Loomba, MD / AASLD 2016 - (07/03/17)
IAS: Fatty Liver in HIV+ at IAS - (08/05/17)
EACS: Analysis of the performance of non-invasive markers of steatosis and fibrosis in HIV-monoinfected patients at-risk of NAFLD: Results from the ERANET-HIVERA ECHAM study - (11/15/17)
EACS: How many HIV mono-infected or HBV or HCV co-infected patients with undetectable viremia should be monitored for liver disease severity in the presence of suspect NAFLD? - (12/08/17)
EACS: Non-viral Liver Disease Burden in HIV Mono-infected Individuals: A Prospective Cohort Study - (12/07/17)
Liver disease in the absence of viral hepatitis co-infection is a growing problem in HIV positive individuals. Possible causes include alcohol, non-alcoholic fatty liver disease, and antiretroviral therapy (ART). We aimed to assess the prevalence of clinically significant hepatic fibrosis (CSHF) and associated risk factors in HIV mono-infected individuals with abnormal liver tests.

EASL: Nonalcoholic Steatohepatitis Drug Pipeline Overview - (07/05/17)
--------------------------------
Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis
Anna M. Diehl, M.D., and Christopher Day, M.D. From the Department of Medicine, Duke University, Durham, NC (A.M.D); and Newcastle University Medical School, Newcastle upon Tyne, United Kingdom (C.D.).- N Engl J Med Nov 23 2017
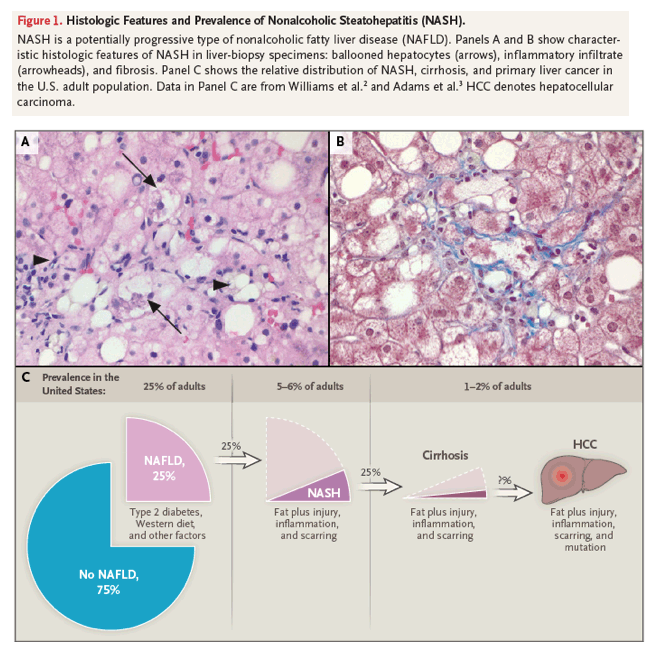
Fatty liver disease is the most common liver disease in the world. About 25% of adults in the United States have fatty livers in the absence of excessive alcohol consumption, a condition termed nonalcoholic fatty liver disease. More than a quarter of adults with nonalcoholic fatty liver disease are presumed to have nonalcoholic steatohepatitis on the basis of elevated serum aminotransferase levels and an absence of other identifiable causes of liver injury.1 A definitive diagnosis of nonalcoholic steatohepatitis is currently based on histologic evidence not only of fat accumulation (steatosis) in hepatocytes but also of liver-cell injury and death and accumulation of inflammatory cells (Figure 1A and 1B). Because livers with nonalcoholic steatohepatitis are more damaged than livers with isolated steatosis, nonalcoholic steatohepatitis is more likely than isolated steatosis to lead to progressive liver fibrosis and eventual liver-related illness and death2-6 (Figure 1C). This review focuses on our understanding of the epidemiology and pathogenesis of nonalcoholic steatohepatitis, which underpins practice guidelines and drug development for this life-threatening liver disease.
Epidemiologic Features
Nonalcoholic steatohepatitis is strongly associated with overweight or obesity and the metabolic syndrome. A recent analysis of studies involving more than 8.5 million persons from 22 countries showed that more than 80% of patients with nonalcoholic steatohepatitis are overweight or obese, 72% have dyslipidemia, and 44% have received a diagnosis of type 2 diabetes mellitus.1 This information supports the concept that nonalcoholic steatohepatitis is the hepatic correlate of the metabolic syndrome, a systemic disorder of energy homeostasis that often accompanies visceral adiposity. Unlike isolated hepatic steatosis, nonalcoholic steatohepatitis is strongly associated with liver fibrosis (scarring), according to liver-biopsy series.4-6 Indeed, these studies show that some level of fibrosis is typical in nonalcoholic steatohepatitis and that liver fibrosis is advanced (defined histologically as fibrosis stage F2 or higher, on a scale ranging from F0 to F4 as follows: no fibrosis [F0], portal fibrosis without septa [F1], portal fibrosis with few septa [F2], bridging septa between central and portal veins [F3], and cirrhosis [F4]) in at least a quarter of patients at diagnosis. Liver fibrosis among patients with nonalcoholic steatohepatitis is to be expected, since wound healing involves fibrosis and patients with nonalcoholic steatohepatitis have greater liver injury than those with isolated steatosis.7
Nonalcoholic steatohepatitis is a dynamic condition that can regress to isolated steatosis, smolder at a relatively constant level of activity, or cause progressive fibrosis that leads to cirrhosis (F4 fibrosis). Natural history studies indicate that the severity of liver fibrosis is the only histologic measure that independently predicts liver-related illness, liver transplantation, and liver-related death in patients with nonalcoholic fatty liver disease.5,6 Analyses of sequential liver-biopsy specimens in patient cohorts indicate that liver fibrosis progresses at a rate of approximately one stage per decade, suggesting that F2 fibrosis will progress to cirrhosis within 20 years. However, rates of fibrosis progression (and regression) vary considerably among individual patients and may not be linear over time even in a given person. In some cases, for example, fibrosis quickly diminishes as nonalcoholic steatohepatitis improves, whereas in others, scarring persists or worsens even after nonalcoholic steatohepatitis has resolved.8
Progressive fibrosis in patients with nonalcoholic steatohepatitis has critical clinical implications because advanced fibrosis reflects repeated futile efforts to regenerate healthy liver architecture, and defective liver regeneration increases the risk of cirrhosis and primary liver cancer.7 Cirrhosis related to nonalcoholic steatohepatitis will develop in approximately 2% of American adults at some point in their lives. This information is derived from fibrosis-progression rates among patients with nonalcoholic fatty liver disease, as well as evidence that approximately 6% of the adult population in the United States has nonalcoholic steatohepatitis and that approximately 25% of affected patients have at least F2fibrosis at the time of diagnosis.1 Indeed, nonalcoholic steatohepatitis will most likely be the top reason for liver transplantation in the United States by 2020.9,10
Nonalcoholic steatohepatitis is also fueling the rising incidence and prevalence of primary liver cancer in many countries, including the United States.11 The incidence of hepatocellular carcinoma is at least 1 to 2% per year among patients with cirrhosis related to nonalcoholic steatohepatitis12; primary liver cancers can also develop in patients with noncirrhotic nonalcoholic steatohepatitis.12,13
In addition, hepatic steatosis and nonalcoholic steatohepatitis occur in children. As with adults, nonalcoholic steatohepatitis in children is strongly associated with obesity. A study of more than 250,000 Danish children showed that childhood obesity increased the risk of hepatocellular carcinoma in adulthood,14 prompting concern that the childhood obesity epidemic in the United States may spawn an epidemic of chronic liver disease, cirrhosis, and liver cancer that will haunt the U.S. population for decades to come. The economic burden of nonalcoholic fatty liver disease in the United States is already enormous, with a current estimate of more than $100 billion in annual direct medical costs, much of which is attributable to nonalcoholic steatohepatitis and its sequelae.15
Pathogenesis
Nonalcoholic steatohepatitis always develops in the context of hepatic steatosis, but isolated steatosis is three or four times as prevalent as nonalcoholic steatohepatitis.1,15 Furthermore, the presence of nonalcoholic steatohepatitis does not correlate with the severity of steatosis as assessed on the basis of liver biopsy or current imaging techniques.6 Since these tests mainly capture hepatic triglyceride content and triglycerides themselves are not directly hepatotoxic, it is presumed that hepatocyte injury is inflicted by toxic triglyceride precursors or products of triglyceride metabolism.16 Evidence that multiple lipid intermediates are cytotoxic, combined with the numerous factors that might enhance or reduce vulnerability to each of these toxic moieties, suggests that nonalcoholic steatohepatitis may be the common manifestation of diverse disease processes.8 Characterizing these processes, developing approaches to detect their presence and intensity, and determining whether one or more pathogenic mechanisms predominate have become major foci for research because such knowledge is necessary for the development of effective interventions for preventing and treating nonalcoholic steatohepatitis.
Hepatocyte injury and death are key features that differentiate nonalcoholic steatohepatitis from isolated steatosis.4 Whether hepatocyte injury is the primary cause or a secondary consequence of liver inflammation is debatable. Both are likely to be relevant to the pathogenesis of nonalcoholic steatohepatitis because injured hepatocytes release factors promoting the accumulation of immune cells that produce hepatotoxic substances and incite further injury and inflammation. Conversely, factors that promote inflammation (e.g., visceral adiposity, the metabolic syndrome, changes in intestinal microbiota, and circadian-rhythm disruption) increase hepatocyte exposure to cytokines, gut-derived products, and other inflammatory mediators that are hepatotoxic.17,18
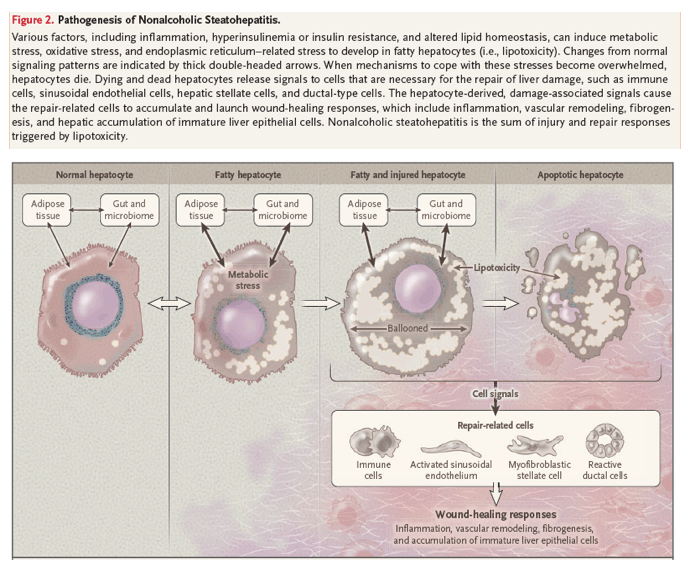
Injured hepatocytes induce stress responses, which limit damage to their vital organelles. However, these adaptations may inadvertently increase vulnerability to other stressors or may simply be insufficient to abort signaling cascades that result in cell death.19,20 Dying hepatocytes, in turn, trigger regenerative responses, promoting the replacement of dead hepatocytes. For example, dying hepatocytes produce morphogens that enrich injured livers with regenerative cell types that are not abundant in healthy livers, such as myofibroblasts, immune cells, and liver-cell progenitors.21,22 An orderly progression of the consequent wound-healing responses results in successful regeneration. However, the wound-healing process stalls during tissue reconstruction when the injury is repetitive or repair becomes dysregulated. Over time, futile regenerative responses promote progressive scarring, which leads to cirrhosis, and perpetuate the stimulus for neoplasia, increasing the risk of liver cancer7 (Figure 2). Histologic assessment provides a snapshot of liver injury and repair responses at the time of biopsy and helps assign patients with nonalcoholic fatty liver disease to prognostic groups4; patients with progressive fibrosis are losing the wound-healing battle and thus may need help to recover.
A number of inherited and environmental factors promote the pathogenesis of nonalcoholic steatohepatitis. Information about some of the pathogenic factors that are becoming diagnostic or therapeutic targets is summarized below.
Genetic Factors
Genomewide association studies indicate that polymorphisms in patatin-like phospholipase domain-containing 3 (PNPLA3) and transmembrane 6 superfamily, member 2 (TM6SF2) promote the development of nonalcoholic steatohepatitis and related liver damage (i.e., cirrhosis, primary liver cancer, or both).23-26 PNPLA3 encodes adiponutrin, a lipase that regulates both triglyceride and retinoid metabolism. PNPLA3 polymorphisms (e.g., I148M) are strongly associated with hepatic steatosis, steatohepatitis, fibrosis, and cancer. The I148M polymorphism also promotes liver damage caused by alcohol-induced fatty liver disease and chronic hepatitis C, an infection that promotes hepatic steatosis. The prevalence of pathologic PNPLA3 polymorphisms differs among ethnic groups, and these differences generally parallel those for nonalcoholic steatohepatitis and its sequelae: both the polymorphisms and the disorders are most prevalent in Asian and native American populations, are less common in whites of Northern European ancestry, and are least common in blacks. However, PNPLA3 I148M is neither sufficient nor necessary to cause nonalcoholic steatohepatitis, cirrhosis, or liver cancer, and the mechanisms through which PNPLA3 aberrancy or adiponutrin dysfunction promotes the pathogenesis of nonalcoholic steatohepatitis are unknown.
TM6SF2 also encodes a protein that regulates hepatocyte lipid content. A TM6SF2 polymorphism that enhances hepatocyte secretion of very-low-density lipoprotein (VLDL) is hepatoprotective but increases the risk of cardiovascular disease, and another TM6SF2 polymorphism, which reduces hepatocyte VLDL secretion, is associated with nonalcoholic steatohepatitis and related liver fibrosis, suggesting that proper trafficking of lipids is necessary to prevent lipotoxicity. Indeed, a deficiency of other proteins required for VLDL secretion also promotes nonalcoholic steatohepatitis and liver fibrosis, but more research is needed to determine how lipid retention incites progressive liver damage. Further research is also necessary to elucidate the role of other gene polymorphisms that are associated with nonalcoholic steatohepatitis-related liver damage.23-26 Defining genetic factors that regulate susceptibility to liver damage is important because studies in twins indicate that heritable factors account for about half the interindividual differences in the prevalence of nonalcoholic steatohepatitis with cirrhosis.27
Environmental Factors
Modifiable risk factors that challenge systemic and hepatic energy homeostasis have been linked to nonalcoholic steatohepatitis, including shift work and alterations in commensal microbiota. Early studies suggested that intestinal microbiota are altered in genetically obese mice with the metabolic syndrome and fatty livers, and these studies linked the abnormal microbiome with hepatic inflammatory signaling that promotes insulin resistance, hepatic steatosis, and nonalcoholic steatohepatitis.36-38 Subsequent work showed that the gut-liver axis has important bidirectional actions that affect health.39-42 The intestinal microbiota influence host susceptibility to obesity, hepatic steatosis, nonalcoholic steatohepatitis, liver fibrosis, and primary liver cancer. Conversely, host factors (e.g., diet composition, adiposity, feeding frequency, and sleep-wake cycles) influence the intestinal microbiota. Early models favored abnormal microbiota as a primary cause of intestinal permeability defects that expose hosts to noxious gut-derived factors (e.g., bacterial lipopolysaccharide, other toll-like receptor ligands, and toxic bile acids). However, more recent studies have shown that when host-generated inflammatory signals are insufficient, the intestinal microbiota are unable to maintain intestinal barrier functions that are hepatoprotective and metabolically favorable.39-42
Whether insights gleaned from studies in animals can inform efforts to prevent or treat nonalcoholic steatohepatitis in humans remains an uncertain, but exciting, possibility. Agents that inhibit recruitment of inflammatory cells, block inflammatory signaling, reduce oxidative stress, and improve insulin sensitivity in preclinical models are being tested in patients with nonalcoholic steatohepatitis. It remains to be determined whether more direct manipulation of the intestinal microbiota with antibiotics, prebiotics, or probiotics can prevent or treat nonalcoholic steatohepatitis. Profiling nonalcoholic steatohepatitis-related changes in intestinal microbiota is an area of active research that is in its infancy but is benefiting from the recent availability of large biobanks of well-characterized human specimens, as well as advances in RNA sequencing and metagenomics.43
Shift work and travel that perturb normal feeding and sleep-wake cycles promote adiposity, the metabolic syndrome, and nonalcoholic fatty liver disease. Studies in mice have revealed that these activities disrupt circadian rhythms at multiple levels (i.e., within the central nervous system, liver, and intestinal microbiome), uncovering integrated mechanisms that normally balance energy supply and demand.44 Transcriptomic analysis of circadian clock-regulated liver genes has shown enrichment with transcripts that control lipid metabolism.45 Prolonged disruption of normal circadian rhythms in mice with fatty livers induces the development of nonalcoholic steatohepatitis by dysregulating cross-talk between two nuclear hormone receptors, farnesoid X receptor (FXR) and constitutive androstane receptor (CAR), leading to suppression of FXR, hepatic accumulation of bile acids, bile acid-induced overactivation of CAR, and eventual CAR-dependent liver injury, fibrosis, and neoplasia.46 Remarkably, the CAR-sensitive liver responses are inhibited by blocking β-adrenergic receptors, which are known mediators of excessive sympathetic nervous system activity caused by disruption of circadian rhythmicity.
These preclinical findings suggest that liver health depends on an integrated network of metabolic responses that coordinate energy supply and demand. Indeed, dysregulation of FXR, CAR, and adrenergic signaling seems to occur in human nonalcoholic steatohepatitis. FXR is part of an exquisite bile acid-sensing system that ensures the optimal size of the bile acid pool. FXR agonists and certain FXR-regulated factors (e.g., fibroblast growth factors 19 and 21) are being evaluated as treatments for human nonalcoholic steatohepatitis. CAR controls hepatic metabolism of xenobiotics and environmental toxins; its activation is required for the carcinogenic actions of phenobarbital.47 Environmental toxins promote both human liver cancer and nonalcoholic steatohepatitis48; thus, inhibiting CAR might be beneficial in patients with nonalcoholic steatohepatitis. Deleting β-adrenergic receptors abrogates hepatic CAR activity in mice,46 and a pilot study has suggested that losartan, an adrenergic antagonist, mitigates nonalcoholic steatohepatitis and fibrosis in humans.49
Diagnostic and Therapeutic Implications
Nonalcoholic steatohepatitis and its deadly sequelae, cirrhosis and liver cancer, are much less prevalent than isolated hepatic steatosis in the general population. This suggests that most persons with fatty livers are able to avoid, constrain, or compensate for stressors that induce nonalcoholic steatohepatitis or drive its progression. Preclinical research has identified a network of interacting factors that determine whether nonalcoholic steatohepatitis develops in fatty livers, as well as whether (and if so, how rapidly) fatal liver damage ensues. This information is beginning to be used to stratify persons with fatty livers into groups that are at higher or lower risk for nonalcoholic steatohepatitis. It also provides a roadmap of potential therapeutic targets, which can guide the development of pharmacotherapy for nonalcoholic steatohepatitis.
Epidemiologic data underscore the importance of differentiating persons with nonalcoholic steatohepatitis from those with isolated hepatic steatosis; only the former group requires resource-intensive, liver-targeted interventions to reduce the risk of liver-related illness and death. Management decisions can be refined by staging liver fibrosis according to its severity. Accurate fibrosis staging is important because biopsy series show that nonalcoholic steatohepatitis is strongly associated with liver fibrosis, and natural history studies show that both all-cause and liver-specific mortality increase once F2 fibrosis has developed. The risk of death from liver disease increases by a factor of 50 to 80 for patients with nonalcoholic steatohepatitis who have F3 or F4 fibrosis, as compared with those who have nonalcoholic steatohepatitis with little or no fibrosis.5,6 These caveats do not indicate, however, that isolated hepatic steatosis is entirely benign, since hepatocyte lipids accumulate in persons who have systemic metabolic stress, and metabolic stress increases the risks of cardiovascular disease and cancer - conditions that themselves warrant preventive and therapeutic interventions.50-52
Dedicated efforts are necessary to identify persons with nonalcoholic steatohepatitis because neither a diagnosis of the disorder nor the severity of associated liver fibrosis is predicted simply on the basis of obesity, insulin resistance, or hepatic steatosis.5,6 However, those conditions should trigger a search for factors that increase the risk of nonalcoholic steatohepatitis, as well as for clues that fibrosis might already be advanced. Overweight or obese persons with the metabolic syndrome, elevated serum aminotransferase levels, and a negative noninvasive workup for other causes of liver disease are likely to have nonalcoholic steatohepatitis. Those who are 45 years of age or older and who have type 2 diabetes are particularly likely to have advanced fibrosis and an increased risk of bad liver outcomes.53 Regardless of risk factors for fibrosis, advanced liver fibrosis should be suspected when even subtle clinical signs of hepatic dysfunction or portal hypertension are detected.
Persons at risk for nonalcoholic steatohepatitis or liver fibrosis should undergo further testing to confirm the clinical suspicion of nonalcoholic steatohepatitis-related liver damage, grade the severity of liver injury, and stage the level of fibrosis, because this information is essential in formulating plans for disease management. Liver biopsy is currently the most widely accepted approach for diagnosing nonalcoholic steatohepatitis and staging liver fibrosis. However, less invasive approaches are being developed that may ultimately permit diagnosis and staging of nonalcoholic steatohepatitis by combining panels of serum biomarkers, new imaging tests that can quantify liver fibrosis, and dynamic tests of liver function.54,55 Genetic screening for polymorphisms that segregate with a high risk of nonalcoholic steatohepatitis, fibrosis, or liver cancer might further improve diagnostic accuracy.24
Accurate diagnosis and staging of nonalcoholic steatohepatitis are essential for the management of this disorder. Nonalcoholic steatohepatitis with no fibrosis (F0) or negligible fibrosis (F1) has an excellent prognosis, and thus intensive follow-up and liver-targeted treatments are not necessary. Currently, lifestyle modifications are the main intervention for nonalcoholic steatohepatitis without fibrosis (Table 1). Adjunctive treatment with vitamin E or pioglitazone, an insulin-sensitizing agent with antiinflammatory actions, might also be considered. A multicenter clinical trial showed that both were superior to placebo for the treatment of nonalcoholic steatohepatitis in nondiabetic patients with mild liver damage and were relatively safe (although pioglitazone often caused weight gain that was not easily lost after treatment was stopped).56That study was not designed to determine the effects on fibrosis progression; hence, the efficacy of either agent for preventing or reversing liver fibrosis is unknown. Generally, persons with nonalcoholic steatohepatitis and F0 or F1 fibrosis are evaluated annually for liver disease progression by means of blood tests and physical examinations. More directed screening for fibrosis progression (e.g., with repeat liver biopsy or newer testing techniques) is often repeated approximately 5 years after diagnosis, but the precise timing of such testing is governed by laboratory and examination results because of the high interindividual variation in rates of fibrosis progression.55

For patients who have more advanced fibrosis (F2 or higher) when nonalcoholic steatohepatitis is diagnosed, management is tailored according to the severity of the fibrosis. Once F3 or F4 fibrosis occurs, the risk of liver-related illness and death increases dramatically, justifying much closer follow-up to detect and treat life-threatening complications of portal hypertension. The same lifestyle modifications that are advised for patients who have nonalcoholic steatohepatitis with F0 or F1 fibrosis are also recommended for patients with more severe fibrosis. Vigorous efforts to determine the most effective methods for management of the metabolic syndrome and for screening for cancer are important, since cardiovascular disease and cancer are the leading causes of death in patients with nonalcoholic steatohepatitis and cirrhosis.
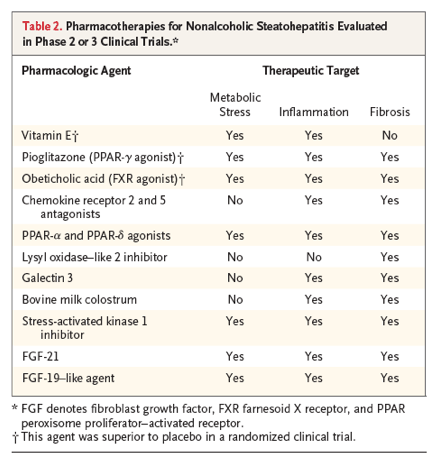
A number of large clinical trials designed to identify effective and safe treatments for nonalcoholic steatohepatitis and liver fibrosis are in progress.57-59 Efforts have focused on ameliorating the three general processes that drive the pathogenesis and progression of nonalcoholic steatohepatitis: metabolic stress, inflammation, and fibrosis. Table 2 lists agents that are in phase 2 or 3 clinical trials. Many of these agents affect more than one of the putative pathogenic targets; a combination of agents that predominantly influence distinct targets is also being considered. Results have been reported for several studies, but no agent has yet received approval by the Food and Drug Administration for the treatment of nonalcoholic steatohepatitis.
A consensus about general principles of treatment for nonalcoholic steatohepatitis is emerging. First, relatively risky and expensive treatments merit consideration in patients with a high risk of bad liver-related outcomes but are not justified in patients at lower risk for disease progression. Second, therapies that increase the risk of cardiovascular disease or cancer should not be used because these disorders are the leading causes of death in patients with nonalcoholic steatohepatitis. Third, the heavy societal burden imposed by liver damage related to nonalcoholic steatohepatitis justifies public health measures aimed at eradicating nonalcoholic steatohepatitis and other metabolic syndrome-related diseases by encouraging healthful eating, facilitating exercise and fitness, limiting shift work, and minimizing exposure to environmental toxins.
Conclusions
Nonalcoholic steatohepatitis, a type of liver damage that is strongly associated with visceral adiposity and the metabolic syndrome, has become a major cause of cirrhosis and liver cancer. The prevalence of nonalcoholic steatohepatitis in the United States approaches that of type 2 diabetes, and annual medical costs directly attributable to nonalcoholic fatty liver disease already exceed $100 billion, much of which is attributable to nonalcoholic steatohepatitis, underscoring the importance of developing interventions to prevent and treat this disease. A number of inherited and environmental factors increase the risk of nonalcoholic steatohepatitis and influence its progression. The pathogenic mechanisms are being unraveled; metabolic stress, inflammation, and fibrosis have been identified as key processes. Pharmacologic agents that target these mechanisms are being investigated in clinical trials.
|
| |
|
|
|
|
|