 |
|
|
| |
Trimming the Fat: Acetyl-CoA Carboxylase
Inhibition for the Management of NAFLD - Editorial
|
| |
| |
Download the PDF here
Download the PDF here
Hepatology 03 August 2018 Norihiro Imai, M.D., Ph.D. David E. Cohen, M.D., Ph.D. - Division of Gastroenterology and Hepatology Joan & Sanford I. Weill Department of Medicine Weill Cornell Medical College New York, NY
Abbreviations
ACC
acetyl-CoA carboxylase
CPT-1
carnitine palmitoyltransferase I
GPAT1
glycerol-3-phosphate acyltransferase 1
SREBP-1c
sterol regulatory element binding protein-1c
VLDL
very-low-density lipoprotein
----------------
SEE ARTICLE ON PAGE 2197 - Acetyl-CoA Carboxylase Inhibition Reverses NAFLD and Hepatic Insulin Resistance but Promotes Hypertriglyceridemia in Rodents
Abstract pdf attached above
Pharmacologic inhibition of acetyl-CoA carboxylase (ACC) enzymes, ACC1 and ACC2, offers an attractive therapeutic strategy for nonalcoholic fatty liver disease (NAFLD) through simultaneous inhibition of fatty acid synthesis and stimulation of fatty acid oxidation. However, the effects of ACC inhibition on hepatic mitochondrial oxidation, anaplerosis, and ketogenesis in vivo are unknown. Here, we evaluated the effect of a liver-directed allosteric inhibitor of ACC1 and ACC2 (Compound 1) on these parameters, as well as glucose and lipid metabolism, in control and diet-induced rodent models of NAFLD. Oral administration of Compound 1 preferentially inhibited ACC enzymatic activity in the liver, reduced hepatic malonyl-CoA levels, and enhanced hepatic ketogenesis by 50%. Furthermore, administration for 6 days to high-fructose-fed rats resulted in a 20% reduction in hepatic de novo lipogenesis. Importantly, long-term treatment (21 days) significantly reduced high-fat sucrose diet-induced hepatic steatosis, protein kinase C epsilon activation, and hepatic insulin resistance. ACCi treatment was associated with a significant increase in plasma triglycerides (approximately 30% to 130%, depending on the length of fasting). ACCi-mediated hypertriglyceridemia could be attributed to approximately a 15% increase in hepatic very low-density lipoprotein production and approximately a 20% reduction in triglyceride clearance by lipoprotein lipase (P ≤ 0.05). At the molecular level, these changes were associated with increases in liver X receptor/sterol response element-binding protein-1 and decreases in peroxisome proliferator-activated receptor-α target activation and could be reversed with fenofibrate co-treatment in a high-fat diet mouse model. Conclusion: Collectively, these studies warrant further investigation into the therapeutic utility of liver-directed ACC inhibition for the treatment of NAFLD and hepatic insulin resistance.
-------------------------------
NAFLD is characterized by the hepatic accumulation of excess fatty acids in the form of triglycerides. Normally, the steady-state concentration of triglycerides in the liver is low. This is because the accrual of fatty acids due to uptake from the plasma and de novo lipogenesis is balanced by β-oxidation within liver mitochondria and by secretion into plasma as very-low-density lipoprotein (VLDL) triglycerides. In the setting of insulin resistance, increased rates of lipolysis within white adipose tissue lead to elevated concentrations of plasma fatty acids and increased rates of uptake into the liver. Insulin resistance also leads to greater rates of hepatic de novo synthesis of fatty acids. Stable isotope studies in human subjects have demonstrated that most (approximately 60%) of excess fatty acids originate from white adipose, but the contribution from de novo lipogenesis is substantial (approximately 25%).1 Because rates of fatty acid oxidation and VLDL secretion are insufficient to compensate, excess fatty acids are sequestered as triglyceride molecules in lipid droplets, which manifest as hepatic steatosis.
Acetyl-CoA carboxylase (ACC) has emerged as a target in the search for NAFLD. ACC catalyzes the first committed step of de novo fatty acids synthesis: the carboxylation of acetyl-CoA to form malonyl-CoA. ACC also regulates fatty acid oxidation because its product malonyl-CoA inhibits carnitine palmitoyltransferase I (CPT-1), the transporter for mitochondrial uptake of fatty acids. Two ACC isoforms exhibit the same enzymatic activity but have distinct locations and metabolic functions. ACC1 is concentrated in the cytosol.
It is highly expressed in lipogenic organs, including liver, adipose tissue, and mammary gland. Cytosolic malonyl-CoA produced by ACC1 is used primarily as substrate for fatty acid biosynthesis. In contrast, ACC2 is localized to mitochondria and is expressed primarily in oxidative tissues, including skeletal and cardiac muscle. Malonyl-CoA produced by ACC2 allosterically inhibits CPT-1 and thereby regulates rates of fatty acid β-oxidation.
Transgenic rodent models support our understanding of the metabolic functions of the two ACC isoforms. Mice globally lacking ACC1 are embryonic lethal, but liver-specific knockout of ACC1 mice are viable and exhibit reduced malonyl-CoA and hepatic triglyceride concentrations.2 Presumably because of its similar enzymatic activity and more than 70% protein identity, ACC2 in hepatocytes can compensate for the liver-specific disruption of ACC1 and support de novo lipogenesis.2 In ACC2 knockout mice, higher rates of fatty acid oxidation are observed in skeletal muscle and heart, which protect against diet-induced obesity and hepatic steatosis.3
The activities of both ACC isoforms are regulated by allosteric and hormonal factors. The lipogenic substrate citrate activates ACC by promoting its polymerization, whereas long chain fatty acyl-CoAs mediate end-product inhibition. Adenosine monophosphate (AMP)-activated protein kinase (AMPK) is the main kinase that controls ACC activity. AMPK is activated when cellular energy is low, leading to phosphorylation and inactivation of ACC. This serves to reduce de novo lipogenesis and enhance fatty acid β-oxidation.
Based on mechanisms by which it controls de novo lipogenesis and fatty acid oxidation, ACC has attracted attention as a potential therapeutic target in NAFLD. Small molecule inhibitors have been identified and tested in rodents and humans. These efforts, including by Goedeke et al.,4 have yielded both promising and unexpected results. ACC inhibitors decrease hepatic triglyceride contents, which is attributable to anticipated effects of reducing de novo lipogenesis and increasing rates of fatty acid oxidation. These findings have now been validated in a phase II trial of a liver-targeted, allosteric inhibitor of ACC1 and ACC2, which demonstrated reduced hepatic steatosis and improved markers of fibrosis.5
Hypertriglyceridemia, an unanticipated response to long-term ACC inhibition, has been observed in both rodents4, 6 and humans.5, 6 Goedeke et al.4 as well as Kim et al.6 have now shed light on this phenomenon as a previously unappreciated mechanistic point of control in triglyceride metabolism. Both groups demonstrated increased rates of VLDL triglyceride secretion from the liver into the plasma. Because hepatic VLDL secretion rates are driven primarily by the availability of triglycerides in the liver, this observation initially seemed counterintuitive. However, a mechanistic analysis revealed decreased hepatic concentrations of polyunsaturated fatty acids, owing to reduced malonyl-CoA that is required for the elongation of essential fatty acids. Polyunsaturated fatty acids are key determinants of gene transcription in the liver. They bind and activate nuclear hormone receptors and reduce the processing of the membrane-bound transcription factor, sterol regulatory element binding protein-1 (SREB1c). Indeed, the reduction of hepatic polyunsaturated fatty acids led to decreases in peroxisome proliferator-activated receptor α (PPARα) activation and increases in SREBP1c activity.
The former downregulates genes that promote fatty acid oxidation, whereas the latter upregulates lipogenic genes. Kim et al.6 further demonstrated that SREBP1c-mediated upregulation of glycerolphosphate acyl-transerase 1 (GPAT1) was critical to the observed increase in VLDL secretion. GPAT1 is a mitochondria-associated enzyme that is rate-determining in triglyceride assembly, and its activity also controls the availability of fatty acids for CPT-1-mediated uptake.7 Knockdown of GPAT1 expression reversed the hypertriglyceridemia by reducing VLDL secretion.6 Goedeke et al.4 also showed that decreased triglyceride clearance in peripheral tissues contributed in part to hypertriglyceridemia. Both groups demonstrated that fibrate drugs, which are synthetic agonists of PPARα, were themselves sufficient to mitigate hypertriglyceridemia secondary to ACC inhibition in rodent models (Fig. 1).
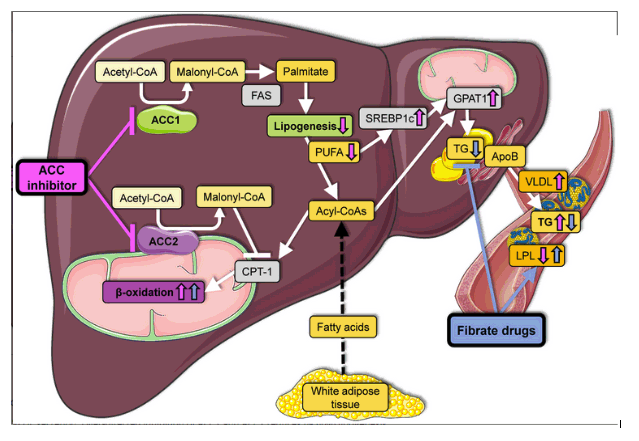
Figure 1
Liver-directed inhibition of ACC enhances fatty acid β-oxidation and reduces de novo lipogenesis but increases VLDL secretion. Liver-directed inhibition of ACC1 and ACC2 reduces de novo lipogenesis and increases fatty acid β-oxidation.4, 6 Despite reductions in triglycerides content in the liver and improvements in insulin sensitivity, liver-directed inhibition of ACC1 and ACC2 leads to a decrease in polyunsaturated fatty acids and an increase in SREBP1c, GPAT1 expression and VLDL secretion, eventually leading to hypertriglyceridemia. Fibrate drugs decrease plasma triglycerides by enhancing fatty acid oxidation in the liver and by promoting lipoprotein lipase-mediated triglyceride clearance from the plasma. Abbreviations: ApoB, apolipoprotein B; FAS, fatty acid synthase; LPL, lipoprotein lipase; PUFA, polyunsaturated fatty acids; and TG, triglycerides.
Cardiovascular disease is the leading cause of morbidity and mortality in patients with NAFLD. Because elevated plasma concentrations of triglyceride may contribute to cardiovascular risk, long-term inhibition of ACC to improve hepatic steatosis in NAFLD may be undermined by this unanticipated, but on-target drug effect. An important unresolved issue is whether ACC-induced hypertriglyceridemia can be fully mitigated by dietary supplementation with polyunsaturated fatty acids or administration fibrate drugs, which can have disparate effects in humans compared with rodents.
Exome-wide association studies have revealed polymorphisms in transmembrane 6 superfamily 2 (TM6SF2) and patatin-like phospholipase domain-containing protein 3 (PNPLA3) that correlate with increased liver fat and risk for diabetes, but reduced plasma triglyceride concentrations and decreased risk of cardiovascular disease.8 PNPLA3 regulates lipid droplet metabolism, whereas TM6SF2 controls VLDL triglyceride secretion. The interactions of ACC inhibitors with these gene variants is unknown.
A related opportunity for ACC inhibitors is in the therapeutic approach to hepatocellular carcinoma. Tumors often exhibit increased rates of fatty acids synthesis to meet increased biosynthetic demands of cell proliferation. The inhibition of lipogenesis by way of ACC has been recognized as a potential modality in cancer chemotherapy. Knockdown of ACC1 reduced cell viability in vitro in human hepatocellular carcinoma cells.9 However, genetic ablation of ACC increased diethylnitrosamine-induced tumorigenesis in the liver,10 apparently because resistance to antioxidant stress served to increase survival of damaged hepatocytes and promote the development of tumor-initiating cells. These divergent findings suggest that appropriate caution will be required when advancing liver-targeted ACC inhibition as a therapeutic strategy for hepatocellular carcinoma.
Assuming that the hypertriglyceridemia is manageable, liver-directed ACC inhibitors appear to hold promise for the management of NAFLD. Experience should reveal whether the efficacy of these inhibitors depends on coexisting allelic variants that modulate hepatic lipid metabolism.
|
| |
|
|
|
|
|