 |
|
|
| |
Effects of Antiretroviral Therapy on Allele-Associated Lipoprotein(a) Levels in HIV
|
| |
| |
Cardiovascular Update CROI 2018 - by Priscilla Hsue MD Professor of Medicine, UCSF - (05/11/18)
Reported by Jules Levin
CROI 2018 March 4-7 Boston MA
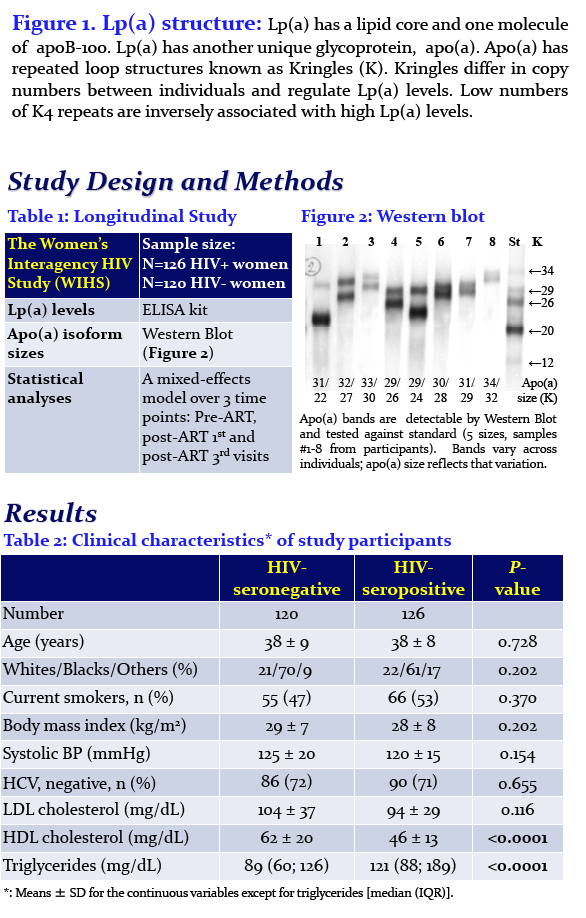
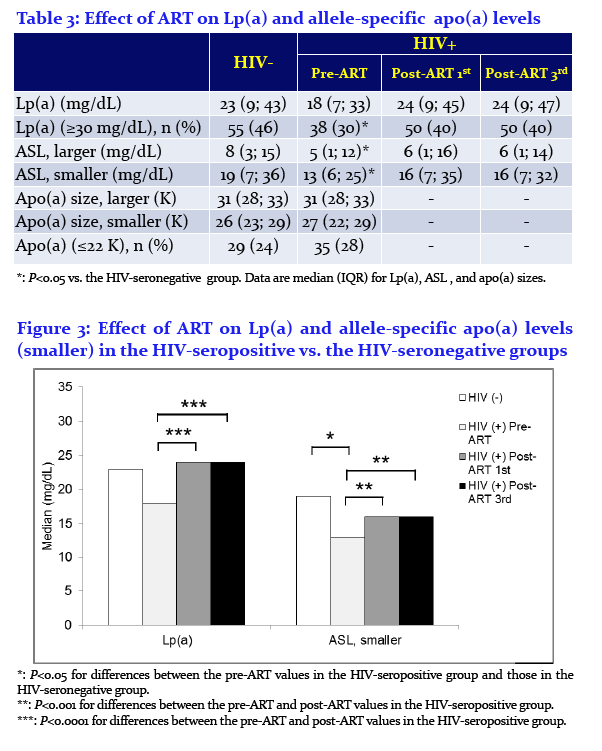
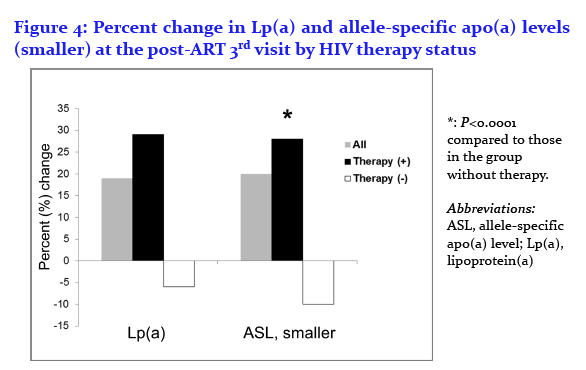
Enkhmaa Byambaa1, Anuurad Erdembileg1, Wei Zhang1, Chin-Shang Li1, Robert Kaplan2, Jason Lazar3, Dan Merenstein4, Roksana Karim5, Brad Aouizerat6, Mardge Cohen7, Kenneth Butler8, Savita Pahwa9, Igho Ofotokun10, Adaora Adimora11, Elizabeth Golub12, and Lars Berglund1
1University of California, 2Albert Einstein College of Medicine, 3SUNY Downstate Medical Center, 4Georgetown University, 5University of Southern California, 6New York University, 7Stroger Hospital, Cook County Bureau of Health Services, Chicago, 8University of Mississippi, 9University of Miami, 10Emory School of Medicine, 11University of North Carolina at Chapel Hill, and 12Johns Hopkins Bloomberg School of Public Health
Medscape
Large Lipoprotein(a) Reductions Needed for Clinical Benefit
Liam Davenport
May 08, 2018
LISBON, Portugal - Levels of lipoprotein(a) [Lp(a)] need to be reduced by substantially more than has been achieved in recent trials to have the same impact on coronary heart disease (CHD) risk as that seen with relatively modest reductions of low-density lipoprotein (LDL) cholesterol, suggests a large-scale analysis of trial data.
In a study that could explain the relatively poor impact of Lp(a) lowering on clinical outcomes to date, the researchers applied the principles of Mendelian genetic inheritance to examine the impact of different Lp(a) reductions on CHD risk in more than 48,000 individuals from five trials.
Presenting the findings here at the European Atherosclerosis Society (EAS) 2018, they showed that an absolute, rather than a relative, reduction in Lp(a) levels of 100 mg/dL would be needed to achieve a similar CHD risk reduction as that seen with a 38.7-mg/dL (1-mmol/L) reduction in LDL cholesterol levels.
Crucially, however, the impact of Lp(a) reduction on CHD risk was not affected by the use of LDL-lowering therapies.
This "would imply that, if you lower LDL, you get a benefit, and if you still have high Lp(a) and you lower Lp(a), you get an additional benefit, independent of the LDL level," study presenter Brian A. Ference, MD, University of Cambridge, United Kingdom, told theheart.org | Medscape Cardiology.
Importantly, those kinds of reductions in Lp(a) levels are achievable with the novel antisense oligonucleotides.
These agents "can lower Lp(a) by 75% to 90%," he said, "so if one started with an Lp(a) levels of, say, 100 to 120 mg/dL, these are the kinds of therapies that...can get an absolute reduction of 100 mg/dL."
Ference noted that other therapies, such as statins, proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, and niacin, "cannot produce those large reductions."
It also important that the right patients are selected for treatment, as the baseline Lp(a) must be high to achieve such large absolute reductions.
"For example, if you start with an Lp(a) of 20 mg/dL and you reduce it by 30%, you're only going to lower it by 6 mg/dL," he said. "That's way too small to get any kind of benefit, so that's why the trials haven't shown any benefit. It's not that lowering Lp(a) doesn't work, it's just that they haven't lowered it enough, and you have to lower it a lot."
However, the fact it has to be lowered a lot points to the patients who are going to benefit, Ference added. "It's only going to be the top 5% of the population, who have really high levels, who can get really large reductions, and therefore get large corresponding reductions in risk."
Valid Interventional Target
Ference began his presentation by pointing out that several recent studies, including meta-analyses and genome-wide association studies, have demonstrated that Lp(a) is a valid interventional target because increased levels are associated with an increased risk for cardiovascular outcomes.
It is notable, however, that the distribution of Lp(a) levels in the population is highly skewed, with 80% of the population having a level less than 50 mg/dL and the remaining 20% a level more than 50 mg/dL.
In addition, recent randomized clinical trials (RCTs) have not been able to show that lowering Lp(a) levels affects risk for CHD.
To determine how much Lp(a) would have to be decreased to achieve a meaningful reduction in CHD risk, the researchers conducted a Mendelian analysis of data from previous trials.
For this, they assumed that, in the same way that treatment is randomly allocated in an RCT, LPA gene variants associated with Lp(a) are naturally randomly allocated in the population, to yield two cohorts: a "treatment" group of individuals with a lower Lp(a) allele and a "usual care" group of those with other alleles.
By examining the incidence of major cardiovascular events between the two groups, it would be possible to quantify the impact of a given difference in Lp(a) levels between them on the risk for CHD events, the researchers say.
The study sample included 20,793 patients with CHD and 27,540 controls from five studies in the CHD Exome+ Consortium, with the findings replicated in up to 189,539 individuals, including 62,240 with CHD.
The team then constructed a genetic risk score from 43 LPA variants associated with Lp(a) mass and measured the overall association between change in Lp(a) levels and the risk for CHD death or nonfatal myocardial infarction.
The analysis showed that the impact of Lp(a) on CHD risk was directly proportional to the absolute change in Lp(a) mass. Specifically, a 10-mg/dL reduction in Lp(a) was associated with an odds ratio of CHD events of 0.942 (95% CI, 0.933 - 0.951; P = 3 × 10-37).
This compares to an odds ratio for a per-38.67-mg/dL (1-mmol/L) reduction in LDL cholesterol levels of 0.511 (95% CI, 0.416 - 0.602; P = 2 × -12).
Further analysis indicated that an 80% reduction in Lp(a) levels would result in a lifelong reduction in a range of CHD outcomes, including coronary disease, myocardial infarction, revascularization, chronic ischemic heart disease, and angina.
The effect of lower Lp(a) levels on CHD was also independent of the effect of statins, ezetimibe, and PCSK9 inhibitors on LDL cholesterol levels.
Ference said that, on the basis of their calculations, a 100-mg/dL reduction in Lp(a) levels would have a similar impact on CHD risk reduction as a 38.1-mg/dL (1-mmol/L) reduction in LDL cholesterol levels, giving an estimated lifelong risk reduction of 44.9% and an estimated short-term risk reduction of 23.7%.
This, he said, not only "explains the failure" of recent cholesteryl ester transfer protein inhibitor, niacin, and PCSK9 trials looking at Lp(a) lowering but also "informs the optimal design of RCTs."
Ference therefore concluded that only individuals with "very elevated Lp(a) levels are likely to derive a large clinically relevant benefit from lowering Lp(a)."
He pointed out that people with Lp(a) levels greater than 180 mg/dL "are likely to have the same lifetime risk of persons with heterozygous familial hypercholesterolemia, but are twofold more prevalent."
He added that because more than 90% of the difference in Lp(a) levels is inherited, "extremely elevated Lp(a) levels may be a new inherited lipid disorder associated with extremely high lifetime risk of CHD."
Catch-22
Approached for comment, M. John Chapman, PhD, Pitié-Salpétrière Hospital, Paris, France, said that the "study is important from the point of view that it clearly confirms a very large evidence base that's accumulated within the last 10 years to the effect that Lp(a) is causal in atherosclerotic cardiovascular disease. Full stop."
The question, he told theheart.org | Medscape Cardiology, "and this is what Brian was trying to approach, is: How high do you need to be at baseline? How high does your overall cardiovascular risk need to be? And, if you target Lp(a), how much do you have to lower it to really see an impact over and above the benefit of LDL reduction from, say, a statin or statin plus PSCK9 inhibitor?"
Chapman points out two "Catch-22s" with this finding. "The first one is how you measure Lp(a), and the difficulty we have is that, around the world, there are several different assays," he said.
He noted that Santica Marcovina, PhD, from the University of Washington, Seattle, "probably has the best assay in the world, but it is exceedingly difficult to purify Lp(a) from anyone, and to make an absolute standard in order to measure, in an unknown plasma sample, the level."
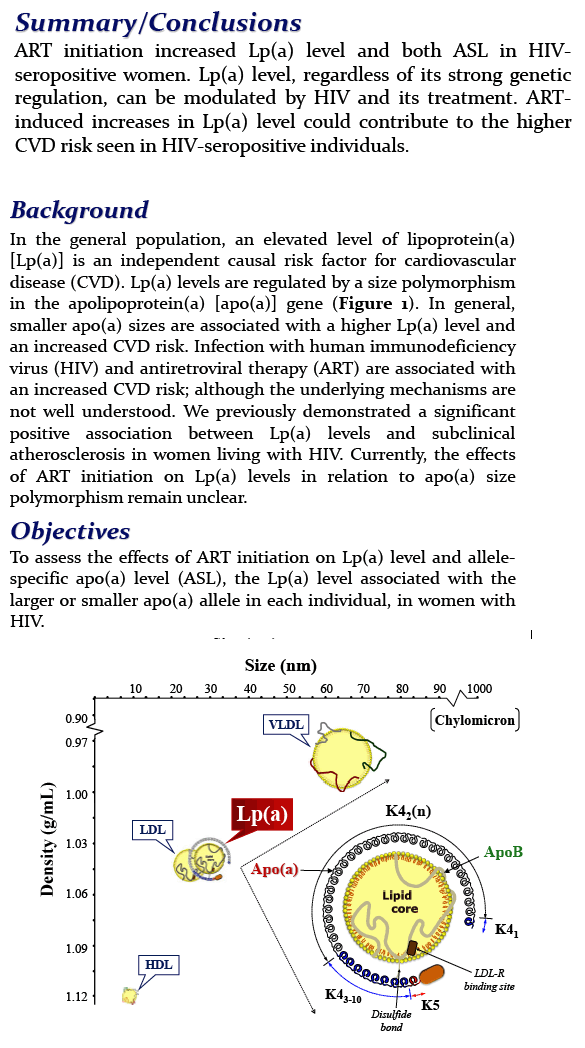
The other issue is that the current study does not speak to the impact of small, or low-molecular-weight, isoforms of Lp(a).
"What you actually need to know is essentially the size of the isoform, and the evidence we have is probably that we don't even need to measure Lp(a) in the blood," he said. "We can probably just do a genetic analysis of the predominant form in a person's blood, because...we know it's a very strong predictor of the level and the risk."
Chapman believes the strength of evidence on the impact of Lp(a) suggests that every patient who has ever had a stroke or myocardial infarction or who has peripheral vascular disease, "in other words, any individual who's in secondary prevention, should be systematically screened either for their Lp(a) level in their plasma or, even better, the genetic isoform."
He concluded that with the ability to assess the Lp(a)-associated risk and the antisense oligonucleotides offering an 80% reduction in Lp(a) levels, "there's light at the end of the tunnel, and it's a big light."
No funding was declared. Ference reports being a consultant for Merck Sharp & Dohme, Amgen, Sanofi/Regeneron, The Medicines Company, Ionis Pharmaceuticals, and CiVi BioPharma; receiving honoraria from Merck Sharp & Dohme, Amgen, Sanofi/Regeneron, The Medicines Company, and Esperion; and receiving research grants from Merck Sharp & Dohme, Amgen, and Esperion.
European Atherosclerosis Society (EAS) 2018. Presented May 6, 2018.
|
| |
|
|
|
|
|