| |
Efficacy, safety, and tolerability of dolutegravir-rilpivirine for the maintenance of virological suppression in adults with HIV-1: phase 3, randomised, non-inferiority SWORD-1 and SWORD-2 studies
|
| |
| |
Download the PDF here
Download the PDF here
Lancet Jan 5 2018 - Josep M Llibre, Chien-Ching Hung, Cynthia Brinson, Francesco Castelli, Pierre-Marie Girard, Lesley P Kahl, Elizabeth A Blair, Kostas Angelis, Brian Wynne, Kati Vandermeulen, Mark Underwood, Kim Smith, Martin Gartland, Michael Aboud
FDA approves first two-drug regimen for certain patients with HIV - Fixed Dose dolutegravir and rilpivirine - FDA News Release - (11/28/17)
JULUCA (dolutegravir and rilpivirine)FDA Prescribing Information - (12/07/17)
Summary
Background
Lifelong HIV antiretroviral therapy (ART) has prompted an interest in two-drug regimens to minimise cumulative drug exposure and toxicities. The safety, tolerability, and efficacy of dolutegravir and rilpivirine suggest potential compatibility and effectiveness as a two-drug regimen. We aimed to investigate this two-drug regimen in a phase 3 study.
Methods
We identically designed SWORD-1 and SWORD-2, which were open-label, parallel-group, multicentre, phase 3, randomised, non-inferiority studies in 12 countries evaluating efficacy and safety of once-daily dolutegravir 50 mg plus rilpivirine 25 mg versus current ART regimen (CAR). We included participants aged 18 years or older who were on first or second ART with stable plasma HIV-1 RNA (viral load <50 copies per mL) for 6 months or longer at screening. We randomly assigned participants (1:1) with stratification by third-agent class, age, and planned participation in a bone mineral density substudy. The primary endpoint was proportion of participants with viral load lower than 50 copies per mL at week 48 among those individuals who received one or more doses of study medication. Investigators monitored adverse events to assess safety. These trials are registered with ClinicalTrials.gov, numbers NCT02429791 (SWORD-1) and NCT02422797 (SWORD-2).
Findings
We screened for participants from April 14, 2015, to Oct 15, 2015, for SWORD-1 and from April 21, 2015, to Sept 25, 2015, for SWORD-2. We randomly assigned 516 participants to dolutegravir-rilpivirine and 512 to continue with CAR. At week 48 (last patient visit was Nov 22, 2016), in the pooled analysis of the intention-to-treat population, 95% of participants had viral loads lower than 50 copies per mL in each group (486 of 513 in the dolutegravir-rilpivirine group vs 485 of 511 in the CAR group), with an adjusted treatment difference of -0•2% (95% CI -3•0 to 2•5) and showed non-inferiority with a predefined margin of -8%. 395 (77%) of 513 participants in the dolutegravir-rilpivirine group and 364 (71%) of 511 participants in the CAR group reported adverse events. The most common adverse events were nasopharyngitis (49 [10%] for dolutegravir-rilpivirine vs 50 [10%] for CAR) and headache (41 [8%] vs 23 [5%]). More participants taking dolutegravir-rilpivirine (17 [3%]) reported adverse events leading to withdrawal than did participants taking CAR (three [<1%]).
Interpretation
Dolutegravir-rilpivirine was non-inferior to CAR over 48 weeks in participants with HIV suppression and showed a safety profile consistent with its components. Results support the use of this two-drug regimen to maintain HIV suppression.
Funding
ViiV Healthcare and Janssen Pharmaceutica NV.
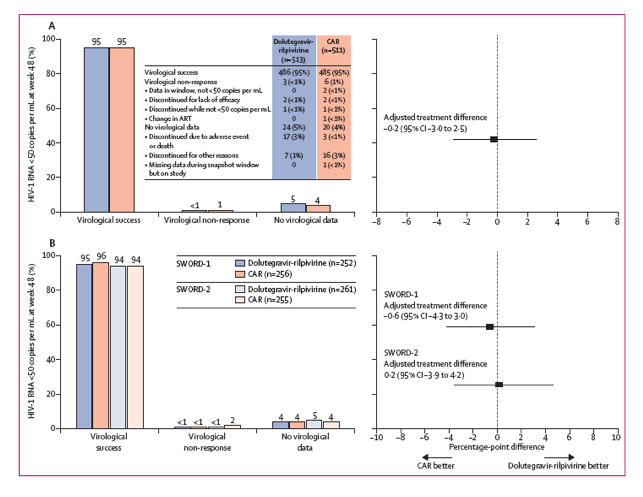
Figure 2: Virological outcomes at week 48 (US Food and Drug Administration snapshot) in the pooled SWORD-1 and SWORD-2 intention-to-treat study population (A) and separated by study (B)
Treatment difference was adjusted for age and baseline third-agent class. CAR=current antiretroviral regimen. ART=antiretroviral therapy.
Introduction
Treatment guidelines for adults with HIV-1 infection recommend first-line and second-line antiretroviral therapy (ART) regimens comprising two nucleoside reverse transcriptase inhibitors (NRTIs) plus a third drug from the boosted protease inhibitor, integrase strand transfer inhibitor (INSTI), or non-NRTI (NNRTI) classes. It has been noted that NRTIs have been associated with long-term negative mitochondrial, renal, cardiovascular, and bone-health effects, prompting clinicians to seek NRTI-sparing options to treat HIV-1 infection.1
Research in context
Evidence before this study
We searched PubMed between April 17 and 27, 2017, for clinical trial publications, cohort studies, and review articles published in English from 2004 to 2017, using combinations, abbreviations, and variations of the search terms "HIV", "antiretroviral therapy", "dolutegravir", "integrase strand transfer inhibitor", "rilpivirine", "non-nucleoside reverse transcriptase inhibitor", "nucleoside reverse transcriptase inhibitor", "dual therapy", "two-drug regimens", and "treatment simplification". We used general Internet searches to acquire relevant practice guidelines and prescribing inserts from governmental, non-governmental, and corporate organisations. The evidence showed that two-drug regimens are being discussed and developed by investigators and clinicians to address emerging polypharmacy, lifelong cumulative drug exposure, ageing, and nucleoside reverse transcriptase inhibitor (NRTI) toxicity issues being faced by patients with HIV-1 infection. The notion for treatment in HIV for almost 20 years has been that three-drug regimens are required to provide adequate virological efficacy and a barrier to emergence of resistance. However, the development and regulatory approval of the potent integrase strand transfer inhibitor dolutegravir has facilitated development of two-drug regimens. The potency, safety, and high-resistance barrier of dolutegravir make it an optimal core drug for an NRTI, protease inhibitor, and booster-sparing two-drug regimen, and the efficacy, safety, and tolerability of rilpivirine suggest compatibility with dolutegravir.
Added value of this study
Results of the SWORD-1 and SWORD-2 studies show that dolutegravir-rilpivirine is non-inferior (predefined -8% non-inferiority margin) to the continuation of triple therapy, consisting of two NRTIs with a third drug belonging to the integrase strand transfer inhibitor, non-nucleoside reverse transcriptase inhibitor, or protease inhibitor classes in maintaining virological suppression through 48 weeks. To date, these studies comprise the largest trial population in which the efficacy and safety of a two-drug regimen have been evaluated. These findings provide a robust demonstration of the suitability of this two-drug regimen, dolutegravir-rilpivirine, for maintenance of HIV-1 suppression in treatment-experienced adults. These studies might also help with the development of new two-drug regimens following careful selection of antiretroviral drugs based on their potency, safety profile, and pharmacokinetic properties.
Implications of all the available evidence
After the early attempts at treatment of HIV infection with combinations of two antiretroviral drugs encountered mixed efficacy and toxicity results, the development and real-world use of this approach in recent years since the advent of combination therapy with three drugs has been sparse. A systematic review of contemporary two-drug regimen studies revealed that the bulk of these studies were insufficiently powered to support robust conclusions regarding efficacy or had no independent confirmation in separate studies. The SWORD studies highlight the potential benefit of two-drug regimens composed of drugs chosen on the basis of their favourable properties (eg, virological efficacy, pharmacokinetic and pharmacodynamic characteristics, and resistance barrier) in highly adherent patients who might need to minimise the number of concomitant medications they are taking. As the population with HIV ages, polypharmacy might become a more important treatment consideration. Treatment simplification strategies with two-drug regimens such as dolutegravir-rilpivirine might become more prominent in treatment decisions.
With combination ART resulting in longer lifespans, ageing patients with HIV are confronted by a constellation of interrelated comorbidities and drug-drug interaction risks.2 Regimens comprising two antiretroviral drugs have been proposed to minimise cumulative drug exposure and reduce risks for long-term drug-related toxicities.3 A systematic review2 of studies on two-drug regimens, from 2000 to 2014, showed mixed results. Most study populations were small, some trials had no active-control groups, or treatment durations were too short to support robust conclusions, and larger randomised trials had no independent confirmation from adequately powered follow-up trials.2 Concerns surrounding two-drug regimens include questions about how they compare with triple therapy in terms of barrier to treatment-emergent resistance and subsequent virological failure.4 Thus, the success of two-drug regimens might depend on choosing component drugs with complementary therapeutic, pharmacokinetic, and pharmacodynamic properties.3
The properties of dolutegravir make it a suitable candidate to investigate in two-drug regimens. Its 14-h plasma half-life and low-to-moderate pharmacokinetic variability between patients support once-daily dosing.5 The drug-drug interaction risks with dolutegravir are low because of its minimal effect on hepatic enzymes and its virological potency without pharmacokinetic boosters.6 The safety and efficacy of dolutegravir have been extensively studied in treatment-naive7, 8 and treatment-experienced9 patients with HIV-1 infection, and it has shown superiority or non-inferiority to comparator regimens, as well as efficacy, regardless of baseline viral load, in each of its phase 3 trials.7, 8, 9 Treatment-emergent resistance has not been reported in any previously ART-naive patient who received dolutegravir in these trials,7, 8 and dolutegravir has been shown to retain activity against most drug-resistance mutations selected by other INSTIs.10
Rilpivirine is an NNRTI with potent virological efficacy and favourable safety profiles when compared with other NNRTIs.11 Its 48-h half-life permits once-daily dosing12 and has shown unchanged virological effectiveness against some resistance mutations selected by other NNRTIs.13 The properties of rilpivirine suggest that it might be compatible with dolutegravir in a once-daily two-drug regimen that blocks viral replication at two stages, thereby enhancing its ability to achieve virological control and suppression. A retrospective cohort study14 of 152 treatment-experienced participants with HIV-1 infection provided preliminary evidence of the safety and efficacy of a once-daily two-drug regimen composed of dolutegravir 50 mg and rilpivirine 25 mg.
Dolutegravir-rilpivirine was found to be well tolerated; 115 (99%) of 116 participants remained free of virological failure, and 105 (91%) remained free of therapeutic failure after 24 weeks. In the SWORD-1 and SWORD-2 studies, we aimed to evaluate the efficacy and safety of dolutegravir-rilpivirine compared with continuation of current ART regimen (CAR) for 48 weeks in a large randomised population with suppressed viral load.
Methods
Study design and participants
The SWORD-1 and SWORD-2 studies are identically designed, 148-week, phase 3, randomised, multicentre (12 countries; appendix), open-label, parallel-group, active-controlled, non-inferiority studies. The studies were done under approval of national, regional, or investigational site ethics committees in accordance with the 2008 Declaration of Helsinki. Summaries of the SWORD-1 and SWORD-2 protocols are available online.15, 16
We included participants who were 18 years or older, were on their first or second ART regimen, and were stably suppressed (viral load <50 copies per mL) for 6 months or longer at screening. Participants had no observed instance of viral load greater than 50 copies per mL in the 6-month period before screening and no more than one instance of viral load greater than 50 copies per mL but lower than 200 copies per mL in the previous period of 6-12 months before screening. Previous ART regimens consisting of two NRTIs plus a third drug (NNRTI, INSTI, or protease inhibitor) were allowed, including pharmacokinetically boosted protease inhibitors or unboosted atazanavir. Enrolment of participants with current or previous exposure to dolutegravir or rilpivirine was limited to about 10%. Key exclusion criteria included any major resistance-associated protease inhibitor, INSTI, NRTI, or NNRTI mutation17 or integrase resistance-associated substitution R263K; severe hepatic impairment (Child-Pugh C); concurrent hepatitis B infection; anticipated need to receive hepatitis C therapy in the first 48 weeks and interferon-based hepatitis C therapy throughout the study; substantial suicidality risk as determined by the site investigator; QT interval corrected according to Bazett's formula of 450 ms or longer; and pregnancy or breastfeeding. We also excluded patients if they switched to a second-line regimen (change of one or more drugs) because of virological failure on the first-line regimen (defined as confirmed plasma HIV-1 RNA ≥400 copies per mL after initial suppression to <50 copies per mL). All participants gave written informed consent before the study began.
Randomisation and masking
We screened potential participants for 14-28 days, and we randomly assigned (1:1) those individuals who met the eligibility criteria to receive dolutegravir 50 mg and rilpivirine 25 mg once daily or continue CAR for 52 weeks. We stratified randomisation by baseline third-agent class (INSTI, NNRTI, or protease inhibitor), age group (< or ≥50 years of age), and planned participation in a bone mineral density substudy (202094 substudy, number NCT02478632). The randomisation and stratification schedule was generated centrally with RandALL NG software. No blinding of the studies was required because they were open label. However, before the week 48 primary analysis, sponsor staff had no access to data summarised by actual randomised treatment.
Procedures
Participants took dolutegravir-rilpivirine at about the same time daily with a meal. We assessed participants during study visits at screening, day 1, weeks 4, 8, 12, 24, 36, and 48, and week 52 (CAR only) or withdrawal. The studies are in progress, with participants having viral load lower than 50 copies per mL at week 48 in the CAR group being switched to dolutegravir-rilpivirine at week 52 to allow time to confirm the week 48 viral load result. We used the Abbott RealTime HIV-1 assay (Abbott Molecular, Des Plaines, IL, USA) to quantify plasma viral load (lower limit of detection of 40 copies per mL).
The confirmed virological withdrawal population included all participants in the intention-to-treat-exposed population (ie, all participants who received at least one dose of study medication) with a viral load of 50 copies per mL or greater and a second, confirmatory viral load of 200 copies per mL or greater. The potential precautionary virological withdrawal population included all participants with consecutive viral loads between 50 copies per mL and 200 copies per mL; these participants were evaluated for possible mitigating circumstances that could have led to these elevations in viral load. If no reason was identified or a participant did not achieve a viral load lower than 50 copies per mL on a third test, we placed the participant in the precautionary virological withdrawal population. For participants who met virological withdrawal criteria, genotypic and phenotypic resistance testing for reverse transcriptase, protease, and integrase was done by Monogram Biosciences (South San Francisco, CA, USA). We analysed CD4 cell counts by flow cytometry.
Potential reasons for withdrawal from the study included drug substitution or dose modification; liver or renal toxicity; QT interval corrected according to Bazett's formula of longer than 500 ms; grade 3 or worse allergic reaction or rash related to study medication; pregnancy; grade 4 clinical adverse event due to study drug; and use of certain prohibited medications.
Adverse events were volunteered by the participant, identified by an investigator questioning the participant, or detected by other means (eg, via central laboratory analysis, suicidality monitoring, or during a physical examination). Investigators noted adverse events as drug related when appropriate.
Outcomes
The primary efficacy endpoint was the proportion of participants in the intention-to-treat population with plasma viral load lower than 50 copies per mL at week 48 using the US Food and Drug Administration (FDA) snapshot algorithm.18 We chose the intention-to-treat population for the primary analysis to provide a conservative approach to evaluation of virological efficacy by including participants who did not have full protocol compliance and were more likely to have virological failure in the snapshot analysis. We did a sensitivity analysis of efficacy in the per-protocol population (intention-to-treat population with the exception of participants with protocol violations that could affect the assessment of antiviral activity or participants who received the correct study medication for <90% of total time on treatment).
Secondary efficacy endpoints were snapshot efficacy at week 24 in the intention-to-treat population; snapshot efficacy at week 48 in the intention-to-treat population in subgroups defined by baseline third-agent class; changes in CD4 T-cell counts at weeks 24 and 48 in the intention-to-treat population; and changes in CD4 T-cell counts at week 48 in subgroups defined by baseline third-agent class. Secondary safety endpoints include incidence and severity of adverse events and laboratory abnormalities over 48 weeks, proportion of participants who discontinued treatment because of adverse events over 48 weeks, change from baseline in fasting lipid concentrations at weeks 24 and 48, and safety assessments based on third-agent treatment class (protease inhibitor, NNRTI, or INSTI). A secondary endpoint regarding virological outcomes was the incidence of observed genotypic and phenotypic resistance to CAR and to dolutegravir or rilpivirine compared with the confirmed virological withdrawal population. Secondary endpoints related to biomarkers included change from baseline in renal, bone, and cardiovascular biomarkers at week 48. Safety was monitored by adverse events, serious adverse events, and other clinical evaluations (appendix).
Endpoints regarding health outcomes were patient-reported outcomes on the HIV Treatment Satisfaction Questionnaire, status version (HIVTSQs); European Quality of Life-5 Dimensions-5 Levels; and the Symptom Distress Module (appendix).
We did an interim futility analysis for the study's independent data monitoring committee (IDMC) to evaluate the efficacy of dolutegravir-rilpivirine about 9 months after the first participant's first study visit with the intention of having roughly 50% of participants reach week 24 in time for the IDMC to review the data before any participants reached the week 48 visit (primary endpoint). The sponsors were masked to this analysis. The IDMC reviewed a futility analysis and monitored the incidence of participants meeting confirmed virological withdrawal criteria by week 36, overall, and within the subset of participants switching from an NNRTI to ensure that participants were not being suboptimally treated. Following this review, the IDMC recommended continuing the study as planned without revision.
Statistical analysis
We designed SWORD-1 and SWORD-2 to ascertain if the antiviral effect of dolutegravir-rilpivirine once daily is non-inferior to CAR at week 48. Two studies were done rather than one larger trial to comply with regulatory guidance and show reproducibility in the efficacy and safety analyses across two international multicentre studies, each enrolling 500 participants or more. The sample size in each study was calculated at 238 participants per group assuming a true 87% response rate (ie, viral load <50 copies per mL at week 48) in each group and a 2•5% one-sided significance level to provide 90% power to infer non-inferiority with a -10% non-inferiority margin chosen to ensure reasonable preservation of treatment effect in the dolutegravir-rilpivirine group. A pooled analysis of all SWORD-1 and SWORD-2 data was predefined, and a non-inferiority margin of -8% was used to provide additional stringency on the analysis of the larger combined population. We calculated the Cochran-Mantel-Haenszel treatment difference (dolutegravir-rilpivirine response rate minus CAR response rate) adjusted for baseline ART third-agent class (ie, INSTI, NNRTI, or protease inhibitor) and age group (ie, <50 years or ≥50 years) to minimise the potential for these variables to confound the analysis. We calculated the treatment difference (dolutegravir-rilpivirine failure rate minus CAR failure rate) adjusted for the same factors as a complementary analysis, with a non-inferiority margin of 4% consistent with updated guidelines from the US FDA.18 We based non-inferiority of the primary endpoint on the difference in response rates. We did all data analysis using SAS software (version 9.1.3 or higher). The trials are registered with ClinicalTrials.gov, numbers NCT02429791 (SWORD-1) and NCT02422797 (SWORD-2).
Role of the funding source
ViiV Healthcare was the financial and regulatory sponsor and participated in designing the trial and collecting, analysing, and interpreting data. Janssen Pharmaceutica NV participated as a partner in the development of the dolutegravir-rilpivirine two-drug regimen. All authors had full access to the data and are responsible for the veracity and completeness of the reported data. The corresponding author had final responsibility for the decision to submit for publication.
Results
We screened for participants from April 14, 2015, to Oct 15, 2015, for SWORD-1 and from April 21, 2015, to Sept 25, 2015, for SWORD-2. The week 48 analysis for both studies included data until Nov 22, 2016. Of 1339 participants screened across both studies, 1028 (77%) were randomly assigned to switch to dolutegravir-rilpivirine (n=516) or continue CAR (n=512; figure 1). The intention-to-treat and safety populations included 1024 participants (figure 1). Participant recruitment ended with about 35 more participants per study than the minimum required for the targeted level of statistical power to allow a sufficient number of participants for a planned substudy of bone mineral density.
Demographic and key baseline clinical characteristics for the intention-to-treat population were well balanced across treatment groups (table 1). Participants were mostly white (819 [80%] of 1024), with a median age of 43 years (range 21-79; table 1) in both groups. At screening, most participants were being treated with ART that included tenofovir disoproxil fumarate or emtricitabine (table 1). Women accounted for 22% of participants in the pooled study population, and most participants were younger than 50 years old (table 1).
59 protocol deviations leading to exclusion from the per-protocol analysis were reported among 56 participants in the dolutegravir-rilpivirine group versus 67 protocol deviations among 58 participants in the CAR group (figure 1). The most frequent protocol deviations were not meeting eligibility criteria and taking prohibited medications. Withdrawals were similar across treatment groups, and the most common reason for withdrawal in the dolutegravir-rilpivirine group was adverse events (figure 1). In the CAR group, the most common reasons were withdrawal of consent and protocol deviation (figure 1). Six participants were withdrawn because of investigators' assessment of lack of efficacy (figure 1).
As recommended by the IDMC, the study progressed through completion of the week 48 primary endpoint analysis. This analysis revealed that 240 (95%) of 252 participants in the SWORD-1 population and 246 (94%) of 261 participants in the SWORD-2 population maintained viral loads of lower than 50 copies per mL after switching to dolutegravir-rilpivirine, compared with 245 of 256 (96% in SWORD-1) and 240 of 255 (94% in SWORD-2) who remained on CAR (figure 2). In the pooled analysis of the intention-to-treat population, 95% of participants maintained viral loads lower than 50 copies per mL in both treatment groups (486 of 513 in the dolutegravir-rilpivirine group vs 485 of 511 in the CAR group) with an adjusted treatment difference of -0•2% (95% CI -3•0 to 2•5; figure 2) that confirmed non-inferiority of dolutegravir-rilpivirine. A sensitivity analysis of the pooled per-protocol population showed an adjusted treatment difference of -0•7% (-3•3 to 1•8), consistent with the primary analysis. Fewer virological failures were reported in the dolutegravir-rilpivirine group than the CAR group (figure 2). Dolutegravir-rilpivirine was non-inferior to CAR in the proportion of participants classified as virological failures (-0•5%, -1•4 to 0•5), with a predefined non-inferiority margin of 4%.
Of the three virological failures in the dolutegravir-rilpivirine group, one met precautionary virological withdrawal criterion, but no study resistance testing was done because the viral load was lower than 200 copies per mL. Two participants met the confirmed virological withdrawal criterion; we did viral resistance testing using findings in one participant with identified non-adherence while receiving dolutegravir-rilpivirine. The data showed the NNRTI resistance-associated substitution K101K/E mixture with no decreased susceptibility to rilpivirine (1•2-fold change); we did not observe any integrase resistance substitutions or decreases in dolutegravir susceptibility. This participant's viral load was 1 059 771 copies per mL at week 36; upon resuming dolutegravir-rilpivirine, the viral load decreased to 1018 copies per mL after 18 days and further decreased to lower than 50 copies per mL at the withdrawal visit (week 45, while still taking dolutegravir-rilpivirine).
Median CD4 cell counts increased from baseline to week 48 (by 28•0 cells per μL, IQR -55•0 to 112•5 in the dolutegravir-rilpivirine group vs by 22•0 cells per μL, -46•0 to 108•0 in the CAR group). Subgroup analyses by age, sex, race, baseline third-agent class, and baseline CD4 cell count gave consistent results to support overall findings (figure 3).
After switching to dolutegravir-rilpivirine, 395 participants (77%) reported at least one adverse event by week 48 compared with 364 participants (71%) who continued with CAR (table 2). The most frequent adverse events were nasopharyngitis, headache, upper respiratory tract infection, diarrhoea, back pain, bronchitis, influenza, and arthralgia, with very few grade 2 or worse events in these categories (table 2, appendix). Adverse events considered to be drug related by the site investigator were reported more frequently in the dolutegravir-rilpivirine group than the CAR group (table 2). Adverse events leading to withdrawal by week 48 were reported in 17 participants (3%) in the dolutegravir-rilpivirine group and three (1%) in the CAR group (table 2).
Neuropsychiatric adverse events were reported more frequently in the dolutegravir-rilpivirine group than in the CAR group (table 2). Insomnia (17 [3%] of 513 in the dolutegravir-rilpivirine group vs ten [2%] of 511 in the CAR group), depression (17 [3%] vs six [1%]), anxiety (11 [2%] vs eight [2%]), and abnormal dreams (six [1%] vs 0) were the most commonly reported neuropsychiatric adverse events at week 48, with steady incremental increases reported in these events at weeks 4, 12, and 24 in the dolutegravir-rilpivirine group (appendix). Few of these events resulted in withdrawal from either group. Other neuropsychiatric adverse events were reported in fewer than 1% of participants in each group. Most psychiatric events were grade 1 or 2 (appendix), with seven participants reporting grade 3 or 4 events (five [1%] of 513 in the dolutegravir-rilpivirine group vs two [<1%] of 511 in the CAR group). Neuropsychiatric adverse events considered to be drug related by the investigator were more frequent in the dolutegravir-rilpivirine group (26 [5%] of 513) than in the CAR group (two [<1%] of 511), with five participants having neuropsychiatric adverse events worse than grade 1 (n=1 grade 4 anxiety, n=1 grade 3 suicidal ideation, and n=2 grade 3 depression in the dolutegravir-rilpivirine group vs n=1 grade 4 suicidal attempt in the CAR group).
Compared with continuing CAR, switching to dolutegravir-rilpivirine had no effect on serum concentrations of total cholesterol, HDL and LDL cholesterol, and triglycerides and no effect on the ratio of total to HDL cholesterol (figure 4). The decrease in serum concentrations of bone-turnover biomarkers, including bone-specific alkaline phosphatase, osteocalcin, procollagen type 1 N-terminal propeptide, and type 1 collagen cross-linked C-telopeptide from baseline to week 48 in the dolutegravir-rilpivirine group differed from the change in the CAR group (figure 4). No consistent pattern of change from baseline to week 48 or differentiation between the dolutegravir-rilpivirine group and the CAR group was observed for the inflammatory or cardiovascular biomarkers: interleukin-6, C-reactive protein, soluble vascular cell adhesion molecule-1, soluble CD14, soluble CD163, fatty acid binding protein-2, and d-dimer (data not shown). A change in fasting glucose concentrations was observed in both treatment groups (median 0•00 mmol/L [IQR -0•30 to 0•40] in the dolutegravir-rilpivirine group vs 0•20 mmol/L [-0•10 to 0•50] in the CAR group; median baseline value for each group was 5•00 mmol/L). Greater decreases were observed in urine retinol-binding protein and urine β-2-microglobulin in the dolutegravir-rilpivirine group than in the CAR group; and no change from baseline was observed at week 48 in serum cystatin C or estimated glomerular filtration rate (using cystatin C) in either group (irrespective of receiving tenofovir disoproxil fumarate at baseline; appendix).
At baseline, similar HIVTSQs total scores were noted in the dolutegravir-rilpivirine group (mean 54•4 [SD 6•4]) and the CAR group (53•9 [6•6]). Greater improvements in HIVTSQs total score from baseline to week 48 were observed in the dolutegravir-rilpivirine group (increase of 1•5; 55•9 [7•0]) than in the CAR group (increase of 0•4; 54•3 [6•0]). Baseline HIVTSQs Lifestyle/Ease subscores were 27•5 [3•2] for the dolutegravir-rilpivirine group and 27•2 [3•3] for the CAR group, with slightly greater improvements at week 48 observed in the dolutegravir-rilpivirine group (increase of 0•8; 28•3 [3•0]) compared with the CAR group (increase of 0•1; 27•3 [3•7]; p<0•0001). No difference was observed in mean change from baseline in HIVTSQs General Satisfaction/Clinical subscores between treatment groups. We observed a significant reduction in mean (SD) Symptom Bother Score in the dolutegravir-rilpivirine group (9•6 [10•0] at baseline vs 8•2 [8•1] at week 48; change from baseline -1•4) compared with the CAR group (11•0 [11•2] at baseline vs 10•3 [9•2] at week 48; change from baseline -0•7; p=0•014). High, stable patient-reported treatment adherence rates were noted in both groups (mean 97•85% [SD 4•22] in the dolutegravir-rilpivirine group vs 98•30% [3•91] in the CAR group).
Discussion
To our knowledge, the SWORD trials have enrolled the largest randomised study population for the evaluation of a two-drug regimen so far2 and both the individual SWORD-1 and SWORD-2 studies and the pooled SWORD data analysis showed non-inferiority in the maintenance of virological suppression over 48 weeks following a switch to dolutegravir-rilpivirine compared with continuing CAR. Notably, the non-inferiority margins (-10% for individual studies and -8% for pooled analysis) were more stringent than the -12% non-inferiority margin used in previous studies7, 19, 20, 21 in this therapy area. The pattern of response rates was consistent across subgroups defined by age, sex, race, baseline CD4 cell count, and baseline third-agent class. Only a few virological failures occurred in the pooled SWORD population at week 48, with non-inferiority of dolutegravir-rilpivirine in this measure shown at an FDA-defined margin of 4%.
Only one mixed NNRTI resistance-associated mutation, with no loss in rilpivirine susceptibility and no INSTI genotypic or phenotypic resistance, was identified in one participant taking dolutegravir-rilpivirine who had acknowledged poor adherence before the elevated viral load result. Nevertheless, this participant was able to achieve viral load lower than 50 copies per mL again after re-establishing adherence with dolutegravir-rilpivirine dosing. The single drug-resistant mutation observed (one [<1%] of 513) might reflect the high barrier to resistance of this dolutegravir-based combination and is lower than the low rate of treatment-emergent resistance reported in the SPIRIT trial (four [1%] of 469),20 which studied switching to rilpivirine-emtricitabine-tenofovir disoproxil fumarate from ritonavir-boosted protease inhibitor therapy in participants with HIV suppression. No integrase resistance-associated substitutions were observed. The SWORD study results indicate that dolutegravir-rilpivirine maintained HIV-1 suppression with no increased risk of developing resistance.
An important consideration regarding the balance of adverse event reporting between the treatment groups is that all participants were stable on their current regimen for at least 6 months before screening. Therefore, we expected participants in the CAR group to tolerate continuation of CAR without substantial new adverse events. Similar differences in adverse events leading to withdrawals have been noted in other switch studies in which participants who stayed on their current regimen reported fewer new adverse events than participants in the switch group.20, 22 Accordingly, patients on ART outside of clinical trials tend to switch to and stay on better-tolerated regimens. In the STRIIVING study,22 75% of patients who switched to dolutegravir plus abacavir-lamivudine reported adverse events at week 48, with 4% having adverse events leading to withdrawal by week 24.
Likewise, more discontinuations due to adverse events were reported after both immediate and delayed switch to rilpivirine-emtricitabine-tenofovir disoproxil fumarate relative to the comparator in the SPIRIT study.20 The open-label design of the SWORD studies might also have affected the greater frequency of adverse events in the dolutegravir-rilpivirine groups. The most frequent adverse events considered to be related to dolutegravir-rilpivirine were aligned with adverse events commonly reported with most ART regimens in previous clinical trials. No new or signature drug-related adverse events were observed with this combined therapy that were not already recognised with the use of the individual components, and no increase was seen in overall frequency or severity of drug-related adverse events. This absence of additive adverse reactions is not surprising given the lack of drug interaction between dolutegravir and rilpivirine.23
Most neuropsychiatric adverse events were grade 1 or 2 and not considered to be related to dolutegravir-rilpivirine. These adverse events often occurred in participants with previous history of anxiety, depression, or insomnia. Because participants in the CAR group were accustomed to their study regimen, few neuropsychiatric adverse events were expected in this group. Additionally, the eligibility criteria excluded participants who posed a clinically significant suicidality risk based on history of suicidal behaviour or ideation at baseline. Finally, the incidence of neuropsychiatric adverse events in the dolutegravir-rilpivirine group was consistent with rates described in a review24 of previous dolutegravir trials, further supporting the absence of any additive or synergistic effects of the two-drug regimen.
Tenofovir disoproxil fumarate, a component of most participants' CAR at baseline, is closely associated with bone demineralisation.25 From baseline to week 48, dolutegravir-rilpivirine was associated with larger reductions in bone-turnover biomarkers (ie, specific alkaline phosphatase, procollagen type 1 N-terminal propeptide, osteocalcin, type 1 collagen cross-linked C-telopeptide) compared with the CAR group in which most participants remained on regimens containing tenofovir disoproxil fumarate. Increased bone-turnover biomarkers have been linked to increased bone loss. We anticipated that the decrease in bone-turnover biomarkers would be associated with preservation of bone health and improvement in bone mineral density following the switch to dolutegravir-rilpivirine.
Additionally, we noted a neutral effect on serum lipids in the dolutegravir-rilpivirine group, despite more than 70% of these participants being switched from tenofovir disoproxil fumarate, which has been reported to be a lipid-friendly drug. This outcome might be an important consideration for older patients living with HIV. Moreover, the neutral effect on lipids could reflect the absence of a booster drug in this two-drug regimen, which can also reduce the potential for drug interactions and can be especially important for patients with extensive polypharmacy burdens. The changes in inflammatory or cardiovascular biomarkers from baseline to week 48 were similar, with no consistent pattern to differentiate between the groups, so no loss of inflammatory control was identified following the switch to this two-drug regimen. Assessment of renal tubule function showed a favourable effect of dolutegravir-rilpivirine compared with CAR.
The SWORD studies had some limitations on the assessment of efficacy and safety. The open-label design might induce bias in both physicians and participants based on their knowledge about the treatment. However, blinding the study medication using a double-dummy design would not have been feasible because it would have resulted in an unacceptably high pill burden, possible negative effects on adherence, and the potential to compromise the evaluation of the endpoints regarding health outcomes. A concern with open-label designs is that knowledge of treatment might affect safety reporting such that more adverse events are reported with novel, experimental treatments (ie, dolutegravir-rilpivirine in the SWORD studies) versus control regimens (ie, CAR). Another limitation is that the SWORD studies were not done in lower-income countries, making it unclear if the results would be relevant in such settings. A strength of the SWORD studies was the successful recruitment of commonly under-represented subpopulations, including women and participants aged 50 years or older. A very low and non-inferior virological non-response rate was observed across both treatment groups, and the two-drug regimen showed no increased risk for selection of resistant HIV-1 infection. To date, the only two-drug regimens that have shown non-inferiority in randomised clinical trials have included a boosted protease inhibitor (lopinavir, atazanavir, or darunavir).19, 26 However, the use of ritonavir as a pharmacokinetic enhancer involves an increased risk of pharmacokinetic interactions and various toxicities when used with some protease inhibitors.27
Once-daily oral dolutegravir-rilpivirine would be the first oral two-drug regimen that provides patients with an alternative to guideline-preferred triple-drug regimens, avoids major NRTI toxicities, has limited potential for drug-drug interactions, and does not increase lipid concentrations or inflammatory biomarkers. Endpoints regarding health outcomes indicated that patients maintained similar levels of satisfaction with dolutegravir-rilpivirine relative to previous regimens and did not report more symptoms during treatment with dolutegravir-rilpivirine. Another potential benefit is the possibility that this novel two-drug regimen could be formulated into one of the smallest complete fixed-dose combination tablets available because it requires only 75 mg of active ingredients (dolutegravir 50 mg and rilpivirine 25 mg). Planned secondary analyses of long-term SWORD endpoints will provide important longitudinal data about the safety of dolutegravir-rilpivirine and its ability to prevent virological failure and treatment-emergent resistance for extended periods.
|
|
| |
| |
|
|
|