| |
FDA Approves Darunavir STR: DRV+Cobi+FTC+TAF
|
| |
| |

Today, FDA approved SYMTUZA™ (darunavir 800 mg, cobicistat 150 mg, emtricitabine 200 mg, and tenofovir alafenamide 10 mg) tablets. SYMTUZA is a four-drug combination of darunavir (DRV), a human immunodeficiency virus (HIV-1) protease inhibitor, cobicistat (COBI), a CYP3A inhibitor, and emtricitabine (FTC) and tenofovir alafenamide (TAF), both HIV-1 nucleoside analog reverse transcriptase inhibitors, and is indicated as a complete regimen for the treatment of HIV-1 infection in adults:
• who have no prior antiretroviral treatment history or
• who are virologically suppressed (HIV-1 RNA less than 50 copies per mL) on a stable antiretroviral regimen for at least 6 months and have no known substitutions associated with resistance to darunavir or tenofovir.
DOSAGE AND ADMINISTRATION
Testing Prior to Initiation:
• Prior to or when initiating SYMTUZA, test patients for HBV infection.
• Prior to or when initiating SYMTUZA, and during treatment with SYMTUZA, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus.
Recommended dosage: One tablet taken once daily with food.
Renal Impairment: SYMTUZA is not recommended in patients with estimated creatinine clearance below 30 mL/min.
Hepatic Impairment: SYMTUZA is not recommended in patients with severe hepatic impairment.
Pregnancy: SYMTUZA is not recommended during pregnancy because of substantially lower exposures of darunavir and cobicistat during pregnancy.
SYMTUZA should not be initiated in pregnant individuals. An alternative regimen is recommended for those who become pregnant during therapy with SYMTUZA.
CONTRAINDICATIONS
SYMTUZA is contraindicated with the following co-administered drugs due to the potential for serious and/or life-threatening events or loss of therapeutic effect [see Drug Interactions (7.5)].
• Alpha 1-adrenoreceptor antagonist: alfuzosin
• Antianginal: ranolazine
• Antiarrhythmic: dronedarone
• Anticonvulsants: carbamazepine, phenobarbital, phenytoin
• Anti-gout: colchicine, in patients with renal/and or hepatic impairment
• Antimycobacterial: rifampin
• Antipsychotics: lurasidone, pimozide
• Ergot derivatives, e.g., dihydroergotamine, ergotamine, methylergonovine
• GI motility agent: cisapride
• Herbal product: St. John’s wort (Hypericum perforatum)
• Hepatitis C direct acting antiviral: elbasvir/grazoprevir
• HMG-CoA reductase inhibitors: lovastatin, simvastatin
• PDE-5 inhibitor: sildenafil when used for treatment of pulmonary arterial hypertension
• Sedatives/hypnotics: orally administered midazolam, triazolam
WARNINGS AND PRECAUTIONS
Drug-induced hepatitis (e.g., acute hepatitis, cytolytic hepatitis) including some fatalities can occur with SYMTUZA. Monitor liver function before and during therapy, especially in patients with underlying chronic hepatitis, cirrhosis, or in patients who have pre-treatment elevations of transaminases).
Severe skin reactions, including Stevens-Johnson syndrome, toxic epidermal necrolysis, drug rash with eosinophilia and systemic symptoms, and acute generalized exanthematous pustulosis may occur with SYMTUZA. Discontinue treatment if severe skin reaction develops.
Patients receiving SYMTUZA may develop new onset or exacerbations of immune reconstitution syndrome.
Monitor in patients with a known sulfonamide allergy.
Discontinue treatment in patients who develop symptoms or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity.
Patients receiving SYMTUZA may develop new onset or exacerbation of diabetes mellitus/hyperglycemia and redistribution/accumulation of body fat.
Patients with hemophilia may develop increase bleeding events.
ADVERSE REACTIONS
The most common adverse reactions (all grades, incidence greater than or equal to 2%) were diarrhea, rash, nausea, fatigue, headache, abdominal discomfort, and flatulence.
Adverse Reactions in Adults with No Prior Antiretroviral Treatment History
Most adverse reactions during treatment with SYMTUZA were grade 1 or 2 in severity. One grade 3 reaction was reported and no grade 4 adverse reactions were reported during treatment with SYMTUZA.
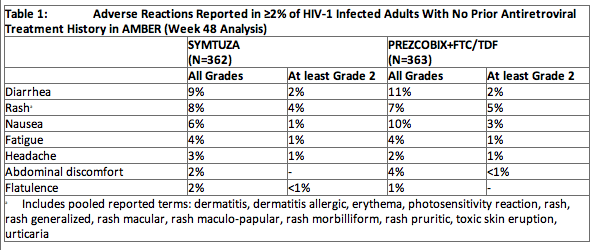
Please refer to the package insert for further details regarding laboratory abnormalities, renal laboratory tests, and bone mineral density outcomes from the two Phase 3 trials.
Adverse Reactions in Virologically-Suppressed Adults
The safety profile of SYMTUZA in virologically-suppressed HIV-1 infected adults is based on Week 48 data from 1,141 subjects in the EMERALD trial, a randomized, open-label, active-controlled trial where 763 subjects with a stable antiretroviral regimen consisting of a boosted protease inhibitor [either darunavir once daily or atazanavir (both boosted with ritonavir or cobicistat), or lopinavir with ritonavir] combined with FTC and TDF switched to SYMTUZA, and 378 subjects who continued their treatment regimen of a boosted protease inhibitor with FTC and TDF. Overall, the safety profile of SYMTUZA in subjects in this study was similar to that in subjects with no prior antiretroviral treatment history. The proportion of subjects who discontinued treatment with SYMTUZA due to adverse events, regardless of severity, was 1%.
CLINICAL STUDIES
Clinical Trial Results in HIV-1 Subjects with no Prior Antiretroviral Treatment History
The efficacy of SYMTUZA in HIV-1 subjects with no prior antiretroviral treatment history was evaluated in the Phase 3 trial TMC114FD2HTX3001 [NCT02431247, (AMBER)] in which subjects were randomized in a 1:1 ratio to receive either SYMTUZA (N=362) or a combination of PREZCOBIX (fixed-dose combination of darunavir and cobicistat) and fixed-dose combination of emtricitabine and tenofovir disoproxil fumarate (FTC/TDF) (N=363) once daily. The median age was 34.0 years (range 18-71), 88.3% were male, 83% White, 11% Black, and 2% Asian. The mean baseline plasma HIV-1 RNA was 4.5 log10 copies/mL (range 1.3-6.7), and 17% had a baseline viral load ≥100,000 copies/mL. The median baseline CD4+ cell count was 453 cells/mm3 (range 38 to 1456 cells/mm3).
Virologic response at 48 weeks of treatment was 91% for SYMTUZA compared to 88% for PREZCOBIX+ FTC/TDF (treatment difference 2.7 (95% CI: -1.6, 7.1). The proportion of subjects with virologic failure was 4% for SYMTUZA and 3% for PREZCOBIX+ FTC/TDF.
The mean increase from baseline in CD4+ cell count at Week 48 was 189 and 174 cells/mm3 in the SYMTUZA and PREZCOBIX + FTC/TDF groups, respectively.
Clinical Trial Results in HIV-1 Virologically-Suppressed Subjects Who Switched to SYMTUZA
Phase 3 trial TMC114IFD3013 [NCT02269917, (EMERALD)] evaluated the efficacy of SYMTUZA in virologically-suppressed (HIV-1 RNA less than 50 copies/mL) HIV-1 infected subjects. Subjects were virologically suppressed for at least 2 months and no more than once had a viral load elevation above 50 HIV-1 RNA copies/mL during the year prior to enrollment. Subjects were on a stable antiretroviral regimen (for at least 6 months), consisting of a boosted protease inhibitor (bPI) [either darunavir once daily or atazanavir (both boosted with ritonavir or cobicistat), or lopinavir with ritonavir] combined with emtricitabine and TDF. Subjects had no history of failure on darunavir treatment and no known or suspected darunavir resistance-associated substitutions. Emtricitabine or tenofovir resistance-associated substitutions were not specifically excluded by the protocol. They either switched to SYMTUZA (N=763) or continued their treatment regimen (N=378) (randomized 2:1). Subjects had a median age of 46 years (range 19-78), 82% were male, 75% White, 21% Black, and 2% Asian. The median baseline CD4+ cell count was 628 cells/mm3(range 111-1921 cells/mm3). Overall, 15% (N=169) of subjects had prior virologic failure. Seven subjects had archived tenofovir resistance-associated substitutions and 53 subjects had archived emtricitabine resistance-associated substitutions, mainly at reverse transcriptase position M184. All of these subjects with emtricitabine resistance-associated substitutions had HIV-1 RNA<50 copies/mL at Week 48 (N=50) or at the last on-treatment viral load (N=3).
Proportion of subjects with HIV-1 RNA > 50 copies/mL at Week 48 was 0.8% for SYMTUZA and 0.5% for bPI+FTC/TDF (treatment difference 0.3 (95% CI: -0.7, 1.2).
The mean increase from baseline in CD4+ cell count at Week 48 was 20 cells/mm3 in subjects who switched to SYMTUZA and 8 cells/mm3 in subjects who stayed on their baseline PI + FTC/TDF.
The updated label will soon be available at drugs@fda or DailyMed
Kimberly Struble
Division of Antiviral Products
Food and Drug Administration
Elizabeth Thompson
Division of Antiviral Products
Food and Drug Administration
Michael Stanfield Jr.
Division of Antiviral Products
Food and Drug Administration
|
|
| |
| |
|
|
|