| |
Dolutegravir plus lamivudine versus dolutegravir plus tenofovir disoproxil fumarate and emtricitabine in antiretroviral-naive adults with HIV-1 infection (GEMINI-1 and GEMINI-2): week 48 results from two multicentre, double-blind, randomised, non-inferiority, phase 3 trials
|
| |
| |
Download the PDF here
November 09, 2018 Lancet
Summary
Background
Effective two-drug regimens could decrease long-term drug exposure and toxicity with HIV-1 antiretroviral therapy (ART). We therefore aimed to evaluate the efficacy and safety of a two-drug regimen compared with a three-drug region for the treatment of HIV-1 infection in ART-naive adults
Methods
We conducted two identically designed, multicentre, double-blind, randomised, non-inferiority, phase 3 trials: GEMINI-1 and GEMINI-2. Both studies were done at 192 centres in 21 countries. We included participants (≥18 years) with HIV-1 infection and a screening HIV-1 RNA of 500 000 copies per mL or less, and who were naive to ART. We randomly assigned participants (1:1) to receive a once-daily two-drug regimen of dolutegravir (50 mg) plus lamivudine (300 mg) or a once-daily three-drug regimen of dolutegravir (50 mg) plus tenofovir disoproxil fumarate (300 mg) and emtricitabine (200 mg). Both regimen drugs were administered orally. We masked participants and investigators to treatment assignment: dolutegravir was administered as single-entity tablets (similar to its commercial formulation, except with a different film colour), and lamivudine tablets and tenofovir disoproxil fumarate and emtricitabine tablets were over-encapsulated to visually match each other. Primary endpoint was the proportion of participants with HIV-1 RNA of less than 50 copies per mL at week 48 in the intention-to-treat-exposed population, using the Snapshot algorithm and a non-inferiority margin of -10%. Safety analyses were done on the safety population. GEMINI-1 and GEMINI-2 are registered with ClinicalTrials.gov, numbers NCT02831673 and NCT02831764, respectively.
Findings
Between July 18, 2016, and March 31, 2017, 1441 participants across both studies were randomly assigned to receive either the two-drug regimen (n=719) or three-drug regimen (n=722). At week 48 in the GEMINI-1 intention-to-treat-exposed population, 320 (90%) of 356 participants receiving the two-drug regimen and 332 (93%) of 358 receiving the three-drug regimen achieved plasma HIV-1 RNA of less than 50 copies per mL (adjusted treatment difference -2⋅6%, 95% CI -6⋅7 to 1⋅5); in GEMINI-2, 335 (93%) of 360 in the two-drug regimen and 337 (94%) of 359 in the three-drug regimen achieved HIV-1 RNA of less than 50 copies per mL (adjusted treatment difference -0⋅7%, 95% CI -4⋅3 to 2⋅9), showing non-inferiority at a -10% margin in both studies (pooled analysis: 655 [91%] of 716 in the two-drug regimen vs 669 [93%] of 717 in the three-drug regimen; adjusted treatment difference -1⋅7%, 95% CI -4⋅4 to 1⋅1). Numerically, more drug-related adverse events occurred with the three-drug regimen than with the two-drug regimen (169 [24%] of 717 vs 126 [18%] of 716); few participants discontinued because of adverse events (16 [2%] in the three-drug regimen and 15 [2%] in the two-drug regimen). Two deaths were reported in the two-drug regimen group of GEMINI-2, but neither was considered to be related to the study medication.
Interpretation
The non-inferior efficacy and similar tolerability profile of dolutegravir plus lamivudine to a guideline-recommended three-drug regimen at 48 weeks in ART-naive adults supports its use as initial therapy for patients with HIV-1 infection.
Funding
ViiV Healthcare.
Discussion
The GEMINI studies showed non-inferior virological efficacy of a two-drug regimen versus a recommended three-drug regimen in the first fully powered, randomised, controlled studies of dolutegravir plus lamivudine in an ART-naive study population. Both regimens were associated with low numbers of confirmed virological withdrawal through week 48; and in participants who met this criterion, neither regimen was associated with emergence of any mutations conferring resistance to integrase strand transfer inhibitors or nucleoside reverse transcriptase inhibitors. Virological suppression rates at week 48 as shown in the GEMINI studies are similar to those observed in other dolutegravir combination studies in ART-naive participants at the week 48 time point. 11, 12, 14 The rapid reduction in viral load, as supported by the percentage of participants who achieved viral suppression at week 4 (72% in the two-drug regimen and 70% in the three-drug regimen) and the rapid median time to viral suppression (29 days), demonstrated that the initial antiviral potency of dolutegravir plus lamivudine is similar to that of the three-drug regimen and consistent with previous reports of dolutegravir plus abacavir and lamivudine from the SINGLE study.23
The GEMINI studies confirm key virological findings of the ACTG A5353 and PADDLE studies. 15, 17 Additionally, the A5353 study showed no difference between the low (≤100 000 copies per mL) and high (>100 000 copies per mL) baseline viral load strata in participants with plasma HIV-1 RNA of less than 50 copies per mL using the US FDA Snapshot algorithm at week 24 (90% for the low viral load strata and 89% for the high viral load strata).17
Similarly, comparable virological efficacy was observed in subgroups stratified by baseline viral load (≤100 000 or >100 000 copies per mL) in the present studies. These results indicate that the two-drug regimen can suppress even high viral loads in ART-naive patients. Although the study aimed to enrol individuals with HIV-1 RNA of less than 500 000 copies per mL, 28 (2%) of 1433 participants had a baseline viral load of 500 000 or more copies per mL (after screening viral load <500 000 copies per mL).
A lower Snapshot response in the two-drug regimen group than in the three-drug regimen group was observed in the subgroup of participants with baseline CD4+ count of 200 cells per μL or less. Most reasons for Snapshot failures (participants who did not have HIV-1 RNA <50 copies per mL at week 48) for this subgroup were unrelated to efficacy or treatment failure, and, as such, the lower response in the participants with lower baseline CD4+ cell counts in the two-drug regimen group should be interpreted with caution.
One perceived risk of treatment with a two-drug regimen compared with a three-drug regimen is greater frequency of drug resistance development in the case of virological failure. In the ACTG A5353 study, three (2%) of 120 participants treated with dolutegravir plus lamivudine had virological failure, and emergence of mutations conferring resistance to treatment was detected in two of the participants (Val106Ile in one patient and Met184Val and Arg263Arg/Leu in the other). 17 However, in the GEMINI studies, ten (1%) of 1433 participants had confirmed virological withdrawal criteria and underwent resistance testing. No emergence of mutations conferring resistance to integrase strand transfer inhibitors or nucleoside reverse transcriptase inhibitors was detected for any participants meeting confirmed virological withdrawal criteria, suggesting there is no increased risk of resistance after virological failure on dolutegravir plus lamivudine compared with a three-drug regimen containing dolutegravir.
Overall safety and tolerability profiles at week 48 were similar between the two regimens, which was expected as dolutegravir was used as the core agent in both regimens. Moreover, the adverse event profile of the two-drug regimen was similar to the recognised profiles of dolutegravir and lamivudine, components of standard-of-care regimens, 24, 25 with no unexpected tolerability or safety findings. Numerically, fewer drug-related adverse events were observed in the two-drug regimen group than in the three-drug regimen group, which was primarily explained by lower frequencies of grade 1 nausea. Changes in renal and bone biomarkers favoured the two-drug regimen and might be attributed to the inclusion of tenofovir disoproxil fumarate in the three-drug regimen group, which is associated with impaired renal function and bone health. 26, 27 Observed lipid changes in the three-drug regimen group, particularly triglycerides and total cholesterol-to-HDL cholesterol ratio, were also consistent with the known effect of tenofovir disoproxil fumarate on cholesterol 28 and were significantly reduced compared with the two-drug regimen group.
This study has several limitations. Enrolment of predominantly men younger than 50 years as participants in a global study suggests that the study demographics do not fully represent the worldwide population living with HIV-1 infection. The analysis of female participants was limited to those using contraceptives and who were not pregnant when initiating treatment. The exclusion criterion of HIV-1 RNA of more than 500 000 copies per mL did not allow any consideration for clinical use of dolutegravir plus lamivudine in treatment-naive individuals with very high viral loads. The study excluded individuals with evidence of hepatitis B virus infection and resistance mutations. It should be noted that 533 (27%) of those screened were not eligible for the study, and the most common reasons for ineligibility were evidence of pre-existing major viral resistance mutations and baseline viral load of less than 1000 copies per mL or more than 500 000 copies per mL. Use of dolutegravir as the core agent in both regimens might constitute another limitation, because dolutegravir could limit the extent to which these safety conclusions can be extrapolated to comparisons with other regimens. Lastly, follow-up beyond 48 weeks will be required to show the durability of dolutegravir plus lamivudine; for this reason, the study is currently ongoing with planned 96-week and 144-week analyses forthcoming.
Of note, our study shows that, as in the GARDEL 9 and ANDES 8 studies, inclusion of a nucleoside reverse transcriptase inhibitor in a two-drug regimen supported by a potent drug with high genetic barrier seems to be crucial for a successful outcome.
Combined with earlier evidence, including comparisons with other agents evaluated in two-drug regimens, these data suggest that the two-drug regimen of dolutegravir plus lamivudine provides high antiviral potency with a high barrier to resistance and favourable safety and tolerability profiles. The GEMINI studies provide strong data supporting dolutegravir plus lamivudine as an effective, well tolerated option for treatment of patients with HIV-1 infection.
Introduction
Standard-of-care first-line therapy for HIV-1 infection in adults naive to antiretroviral therapy (ART) is a regimen of three antiretroviral agents that includes two nucleoside reverse transcriptase inhibitors and one other drug from either the boosted protease inhibitor, integrase strand transfer inhibitor, or non-nucleoside reverse transcriptase inhibitor classes.1 However, concerns exist regarding the toxicities accruing from cumulative exposure to drugs that need to be taken for life. 2 Thus, two-drug regimens capable of inducing or maintaining virological suppression, or both, while decreasing lifetime cumulative drug exposure and potential long-term toxicities would represent another treatment option for people living with HIV-1 infection.
-----------------------------
Research in context
Evidence before this study
We searched PubMed for publications of clinical trials, cohort studies, and review articles using combinations, abbreviations, and variations of the search terms "HIV", "antiretroviral therapy", "dolutegravir", "integrase strand transfer inhibitor", "lamivudine", "nucleoside reverse transcriptase inhibitor", "dual therapy", "two-drug regimens", "treatment-naive", and "treatment simplification". We also used other public search engines to identify and retrieve relevant practice guidelines, regulatory documents, and package inserts from governments, non-governmental organisations, and companies. We did the searches from May 17, 2018, to May 30, 2018, and included materials published from Jan 1, 1995, to May 30, 2018. We observed from this search an increasing concern regarding the potential consequences of exposing people living with HIV-1 infection to combinations of three or more antiretroviral drugs for the duration of their lives, which have been lengthened because of the success of combination antiretroviral therapy (ART). All ART agents are associated with specific adverse events or other challenges to tolerability and adherence to therapy. Two-drug regimens are being developed to simplify ART and reduce lifelong drug exposure and toxicity compared with current standard of care, which requires a combination of three or more drugs.
Added value of this study
The GEMINI studies provide evidence from two fully powered, phase 3 studies that a two-drug regimen of dolutegravir plus lamivudine is non-inferior at a -10% non-inferiority margin compared with a recommended standard-of-care three-drug regimen (ie, dolutegravir plus tenofovir disoproxil fumarate and emtricitabine) for the treatment of ART-naive, HIV-1-infected participants through 48 weeks. In particular, these studies show that the antiviral activity of dolutegravir plus lamivudine is similar to a dolutegravir-based three-drug regimen in participants with screening HIV-1 RNA of 1000-500 000 copies per mL. There were fewer participants with advanced disease (ie, CD4+ count <200 cells per μL) in the two-drug regimen group who achieved virological response, although this finding was confounded by low sample size and was not associated with a substantially higher frequency of treatment-related discontinuations. Importantly, dolutegravir plus lamivudine was not associated with treatment-emergent mutations, suggesting a high barrier to resistance.
Implications of all the available evidence
These studies are the first to demonstrate the non-inferiority of a two-drug regimen containing dolutegravir in ART-naive individuals. Furthermore, these data are the first to show similar efficacy of any two-drug regimen with a standard integrase strand transfer inhibitor-based three-drug regimen, regardless of baseline viral load. These study results support dolutegravir as an optimal core agent for use in two-drug regimens. The GARDEL study showed non-inferiority of the two-drug regimen of lopinavir plus ritonavir and the ANDES study showed non-inferiority of darunavir plus ritonavir both in combination with lamivudine, suggesting lamivudine is a suitable agent for a two-drug regimen for treatment-naive individuals. The present results show that a two-drug regimen of dolutegravir plus lamivudine can be used for the treatment of HIV-1-infected adults naive to ART with a screening viral load of 500 000 copies per mL or less. As cumulative drug exposure becomes a more important treatment consideration with patients living longer, initial treatment strategies composed of two-drug regimens such as dolutegravir plus lamivudine might become an essential factor in treatment decisions.
--------------------
Early trials between 2000 and 2014 evaluating two-drug regimens in treatment-naive populations yielded inconclusive results, perhaps partly because of small sample sizes, short treatment durations, and limitations of available treatments. 3 The phase 3 MODERN trial (n=797) reported inferior viral suppression with maraviroc plus ritonavir-boosted darunavir compared with a three-drug regimen. 4 The AIDS Clinical Trials Group Study A5142 team found that the virological efficacy of a two-drug regimen comprising efavirenz plus ritonavir-boosted lopinavir (n=250) was similar to that of efavirenz plus two nucleoside reverse transcriptase inhibitors (n=250) through 96 weeks of treatment in ART-naive participants but was associated with increased emergence of drug resistance. 5 In the phase 3 NEAT001/ANRS143 trial (n=805), raltegravir plus ritonavir-boosted darunavir was non-inferior to a three-drug regimen in the overall population of treatment-naive participants. 6 However, the two-drug regimen was inferior to the three-drug regimen in the subgroup of participants with CD4+ counts of less than 200 cells per μL and did not show non-inferiority with the subgroup of participants with baseline HIV-1 RNA of more than 100 000 copies per mL. The results of these studies highlight the need for two-drug regimens to contain agents with potent antiviral activity and high barrier to resistance. The reported efficacy, safety, and tolerability of lamivudine over the past two decades of use make it a good candidate as a two-drug regimen partner. 7 Week 48 results from the ANDES trial (n=145) showed that lamivudine plus ritonavir-boosted darunavir is non-inferior to a ritonavir-boosted darunavir-based three-drug regimen for achieving HIV-1 RNA of less than 50 copies per mL in treatment-naive HIV-1-infected patients. The GARDEL study showed the non-inferior virological efficacy of open-label ritonavir-boosted lopinavir plus lamivudine (n=214) compared with ritonavir-boosted lopinavir plus two nucleoside reverse transcriptase inhibitors (n=202) in ART-naive participants after 48 weeks, although it should be noted that 54% of participants in the three-drug regimen group were taking zidovudine. 9 Results from these two studies suggest that lamivudine is an effective two-drug regimen partner with an antiretroviral agent with a high barrier to resistance. However, these two-drug regimens included ritonavir-boosted protease inhibitors, which are associated with a variety of metabolic adverse reactions and drug-drug interactions, and might negate any anticipated benefit from decreased cumulative toxicity due to reduced drug exposure.10
The phase 3 clinical development of dolutegravir as a component of a three-drug regimen has provided extensive evidence that dolutegravir is a good candidate as a core agent in two-drug regimens, with up to 144 weeks of follow-up data supporting the virological efficacy, favourable tolerability profile, and high barrier to resistance. 11, 12, 13, 14
The two-drug regimen of dolutegravir plus lamivudine was evaluated in the 48-week pilot study PADDLE for the treatment of ART-naive participants.15 By week 8, all 20 participants, including four with baseline HIV-1 RNA of 100 000 copies per mL or more, had achieved a viral load of less than 50 copies per mL; 18 participants maintained virological suppression at week 48, 15 and all 18 participants remained suppressed at week 96. 16
Additionally, the phase 2, single-arm ACTG A5353 study in 120 treatment-naive participants with HIV-1 RNA of less than 500 000 copies per mL treated with dolutegravir plus lamivudine showed that 90% (n=108) achieved HIV-1 RNA of less than 50 copies per mL at week 24, regardless of baseline HIV-1 RNA.
17 Thus, dolutegravir plus lamivudine might constitute a viable combination as a novel two-drug regimen for treatment of HIV-1 infection in ART-naive patients.
The primary objective of the GEMINI-1 and GEMINI-2 studies was to therefore evaluate the efficacy and safety of the two-drug regimen comprising dolutegravir plus lamivudine compared with the three-drug regimen of dolutegravir plus tenofovir disoproxil fumarate and emtricitabine for treatment of HIV-1 infection in ART-naive adults.
Methods
Study design and participants
We did two identically designed, multicentre, double-blind, randomised, non-inferiority, phase 3 trials: GEMINI-1 and GEMINI-2. Both studies were done at 192 centres in 21 countries (appendix). Study protocols were approved by national, regional, or investigational centre ethics committees and institutional review boards in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Guideline for Good Clinical Practice. The study protocols for GEMINI-1 and GEMINI-2 are available online.
We recruited participants who were 18 years or older with HIV-1 infection and naive to ART (defined as ≤10 days of previous therapy with any ART). Entry criteria at study start specified screening viral loads of 1000-100 000 copies per mL but, as permitted per protocol, the upper limit was increased to 500 000 copies per mL during the study after an independent review of data from independently sponsored studies evaluating the two-drug regimen of dolutegravir plus lamivudine. We included women of reproductive potential if they were not pregnant or lactating, and were using approved contraception. We excluded individuals with pre-existing major viral resistance mutations to nucleoside reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors, or protease inhibitors; 18 and active US Centers for Disease Control and Prevention stage 3 HIV disease, 19
except for cutaneous Kaposi's sarcoma and CD4+ cell counts of less than 200 cells per μL. The appendix provides further information about the exclusion criteria. We obtained written informed consent from each participant before initiation of study procedures.
Randomisation and masking
We randomly assigned participants (1:1) to receive a two-drug regimen of dolutegravir plus lamivudine or a three-drug regimen of dolutegravir plus tenofovir disoproxil fumarate and emtricitabine. We screened and stratified participants by HIV-1 RNA (≤100 000 or >100 000 copies per mL) and CD4+ cell count (≤200 or >200 cells per μL). Treatment assignment was done in accordance with a central randomisation schedule generated with SAS (version 9.2). We masked both participants and investigators to treatment assignment until week 96. Some sponsor personnel were unmasked at week 24 to facilitate analysis of interim results for submission to regulatory authorities. Dolutegravir was administered as single-entity tablets similar to its commercial formulation, except with a different film colour. Lamivudine tablets and tenofovir disoproxil fumarate and emtricitabine fixed-dose combination tablets were over-encapsulated to visually match each other.
Procedures
Participants received either a once-daily two-drug regimen of dolutegravir (50 mg) plus lamivudine (300 mg) or a once-daily three-drug regimen of dolutegravir (50 mg) plus tenofovir disoproxil fumarate (300 mg) and emtricitabine (200 mg). Both drug regimens were administered orally. The study comprises a screening period of up to 35 days, a double-blind randomised phase from day 1 to week 96, and an open-label randomised phase from weeks 96 to 148. Study visits for all participants were planned at baseline and at weeks 4, 8, 12, 16, 24, 36, and 48, with additional retest visits at weeks 28 and 52 to confirm viral test results for participants with HIV-1 RNA of 50 copies per mL or more at weeks 24 or 48, respectively. Subsequent study visits every 12 weeks are planned through week 148. This report describes results of the primary analysis at week 48 that was done after the last participant had their week 48 viral load assessment, including a week 52 retest as appropriate, in accordance with the US Food and Drug Administration (FDA) Snapshot algorithm. 20
Participants met confirmed virological withdrawal criteria if a second and consecutive HIV-1 RNA value met any of the following definitions: decrease from baseline in HIV-1 RNA of less than one log10 copies per mL, unless HIV-1 RNA of less than 200 copies per mL, by week 12; confirmed plasma HIV-1 RNA of 200 copies per mL or more at or after week 24; or confirmed rebound (HIV-1 RNA ≥200 copies per mL after confirmed consecutive HIV-1 RNA <200 copies per mL). These participants were discontinued from the study, and a plasma sample from the initial suspected viral load sample was used for genotypic and phenotypic resistance tests. The following prespecified resistance mutations of integrase strand transfer inhibitor were included for determining the emergence of resistance mutations: His51Tyr, Thr66Ala/Ile/Lys, Leu74Met, Glu92Gln/Val/Gly, Gln95Lys, Thr97Ala, Gly118Arg, Phe121Tyr, Glu138Ala/Lys/Asp, Gly140Ala/Cys/Ser, Tyr143Cys/His/Arg/Lys/Ser/Gly/Ala, Pro145Ser, Gln146Pro, Ser147Gly, Gln148His/Lys/Arg, Val151Ile/Leu/Ala, Ser153Phe/Tyr, Asn155His/Ser/Thr, Glu157Gln, Gly163Arg/Lys, Ser230Arg, Arg263Lys, Leu68Val/Ile, Leu74Ile, Glu138Thr, and Gly193Glu. We used the most recent International Antiviral Society-USA guidelines available at the time of the database lock to determine major resistance mutations to nucleoside reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors, and protease inhibitors. 18
In addition to virological efficacy, we assessed adverse events, concomitant medications, and symptom-directed physical examinations at all study visits. We collected plasma for quantitative HIV-1 RNA analysis and storage at all visits, and we used the Abbott RealTime HIV-1 assay (lower limit of quantitation, 40 copies per mL; Abbott Molecular, Des Plaines, IL, USA) for the quantitative analysis. We coded adverse events using MedDRA (version 21.0), and graded the maximum toxicity of adverse events using guidelines from the Division of AIDS (version 2.0). 21
Clinical chemistry values, haematology values, and lymphocyte subsets were assessed at all routine study visits. Testing for fasting lipids and glucose, urinalysis, and assessments of renal and bone biomarkers were done at baseline and weeks 24 and 48. Renal biomarkers assessed were serum and urine creatinine; ratios of urine albumin and protein, retinol-binding protein, and β-2 microglobulin to urine creatinine; serum cystatin C; and estimated glomerular filtration rate, which was calculated using creatinine or cystatin C parameters with the Chronic Kidney Disease Epidemiology Collaboration equation. Bone biomarkers assessed were bone-specific alkaline phosphatase, osteocalcin, procollagen type 1 N-terminal propeptide, and type 1 collagen cross-linked C-telopeptide. Pregnancy tests were done for women of reproductive potential at all study visits. We used the Columbia Suicide-Severity Rating Scale to monitor suicidal ideation and behaviour at all routine study visits starting from day 1.
Changes in health-related quality of life were assessed using the EuroQol-5 Dimensions-5 Levels (EQ-5D-5L) questionnaire 22 at baseline; weeks 4, 24, and 48; or upon study withdrawal. The EQ-5D-5L consists of two distinct scoring components: the health state utility score (potential range of -0⋅281 to 1) and the visual analogue scale (VAS; potential range of 0-100). Higher scores on the EQ-5D-5L indicate better health.
Outcomes
The primary objective of each GEMINI study was to show the non-inferior virological efficacy of the two-drug regimen compared with the three-drug regimen, with the primary endpoint being the proportion of participants with plasma HIV-1 RNA of less than 50 copies per mL at week 48 using the FDA Snapshot algorithm 20 in the intention-to-treat-exposed population. Secondary endpoints included proportion of participants with HIV-1 RNA of less than 50 copies per mL at week 24, time to achieve HIV-1 RNA of less than 50 copies per mL, absolute values and change from baseline to week 48 in CD4+ cell count, disease progression (ie, HIV-associated conditions, AIDS, or death), and incidence of emergence of mutations conferring genotypic and phenotypic resistance to dolutegravir plus lamivudine or tenofovir disoproxil fumarate and emtricitabine in participants meeting criteria for confirmed virological withdrawal. Virological efficacy was also assessed in participant subgroups defined by demographic and baseline disease characteristics, including plasma viral load, CD4+ cell count, hepatitis C virus infection, HIV-1 infection subtype, age, sex, race, and ethnicity. Safety endpoints included incidence and severity of adverse events and proportion of participants who discontinued treatment because of adverse events. Renal and bone biomarkers as well as lipids were monitored by assessing changes from baseline to weeks 24 and 48. A complete list of secondary endpoints is provided in the appendix.
Statistical analysis
Two studies were done to comply with regulatory guidance and to show reproducibility in the efficacy and safety analyses across two fully powered studies. For each study, the targeted sample size of 347 participants per treatment group was based on 90% power, a 2⋅5% one-sided α level, a non-inferiority margin of -10% for virological efficacy (non-inferiority concluded if the lower boundary of the two-sided 95% CI for the difference in response is greater than -10% between the two groups), and assumed true response of 87% for the two-drug regimen and 89% for the three-drug regimen. There was no predefined non-inferiority margin for the pooled analysis. All participants who received one or more doses of study medication were included in the intention-to-treat-exposed population, which was used for the primary efficacy analysis. The safety population included all participants who received one or more doses of study medication and was analysed according to actual treatment received. The per-protocol population comprised all participants in the intention-to-treat-exposed population, except for those with a protocol violation that could affect assessment of antiviral activity; this population was used for sensitivity analyses.
For the primary efficacy endpoint, the proportion of responders at week 48 was analysed using a Cochran-Mantel-Haenszel test stratified by plasma HIV-1 RNA (≤100 000 vs >100 000 copies per mL) and CD4+ cell count (≤200 vs >200 cells per μL) at baseline. Baseline characteristics, proportion of participants who achieved a response by study visit or participant subgroup (using Snapshot algorithm), and adverse events were summarised with descriptive statistics. Protocol-defined serious adverse events included those that resulted in death, disability, incapacity, congenital anomaly, or birth defect; are life-threatening; require hospitalisation or prolongation of existing hospitalisation; are associated with liver injury and impaired liver function (ie, alanine aminotransferase ≥three times the upper limit of normal [ULN] and either total bilirubin ≥two times the ULN or international normalised ratio >1⋅5); or require a medical or surgical intervention to prevent a serious adverse event. Participants who had not met confirmed virological withdrawal criteria and were ongoing in the study, or who had discontinued for reasons other than those related to treatment or lack of efficacy, were censored. Change from baseline to week 48 in CD4+ cell counts was analysed with ANCOVA on the basis of a dataset in which multiple imputations have been drawn from a multivariate normal model with a Markov chain Monte Carlo approach to impute missing observations. The statistical model was adjusted for treatment, baseline CD4+ cell count as a covariate, randomisation strata term baseline plasma HIV-1 RNA (≤100 000 vs >100 000 copies per mL), and baseline CD4+ cell count (≤200 vs >200 cells per μL). Time-to-viral suppression was based on time of first viral load of less than 50 copies per mL estimated with non-parametric Kaplan-Meier method. We used SAS version 9.4 within a LINUX environment for all statistical analyses.
An independent data monitoring committee provided external review of efficacy and safety data, including a futility analysis when approximately half of participants in each study had completed the week 24 study visit. GEMINI-1 and GEMINI-2 are registered with ClinicalTrials.gov, numbers NCT02831673 and NCT02831764, respectively.
Role of the funding source
The funder of the study had a role in the study design, data collection, data analysis, data interpretation, and writing the report. All authors had full access to the data and are responsible for the accuracy and completeness of this report. The corresponding author had final responsibility for the decision to submit for publication.
Results
Participant screening commenced on July 21, 2016, for GEMINI-1, and July 18, 2016, for GEMINI-2. Screening ended on March 28, 2017, for GEMINI-1 and March 31, 2017, for GEMINI-2. The data cutoff date used for the week 48 database freeze was May 22, 2018. Of the 1974 participants screened across both studies, 1441 met eligibility criteria and were randomly assigned to receive either the two-drug regimen (n=719) or three-drug regimen (n=722); 533 were not eligible (figure 1). Eight participants randomly assigned to the two-drug regimen (n=3) or three-drug regimen (n=5) groups did not receive study treatment, leading to 716 participants in the two-drug regimen group and 717 in the three-drug regimen group being included in the primary efficacy and safety analysis (GEMINI-1: 356 participants for the two-drug regimen and 358 for the three-drug regimen; GEMINI-2: 360 for the two drug-regimen and 359 for the three-drug regimen). Key demographic and baseline clinical characteristics were well balanced between the treatment groups in the intention-to-treat-exposed population pooled across both trials (n=1433; table 1; appendix p 4). Participants had a median age of 33 years (range 18-72), with most participants being younger than 50 years (1288 [90%] of 1433), men (1222 [85%]), and white (977 [68%]). Baseline HIV-1 RNA of more than 100 000 copies per mL occurred in 293 (20%) and CD4+ cell count of 200 cells per μL or less occurred in 118 (8%) participants.
In the pooled study population, the numbers of participants who discontinued treatment at the time of the database lock at week 48 were similar between the two-drug regimen (66 [9%] of 719) and three-drug regimen (52 [7%] of 722) groups (figure 1). The most common reasons for study withdrawal were participant lost to follow-up (26 [2%] of 1433), discontinuation due to adverse events (23 [2%]), and withdrawn consent (23 [2%]). Withdrawals due to protocol deviations occurred for ten (1%) participants in the two-drug regimen group compared with six (<1%) in the three-drug regimen group; the most common reasons were due to non-compliance with protocol procedures (eight [<1%] of 1433). Four participants (two in each treatment group) became pregnant during the study.
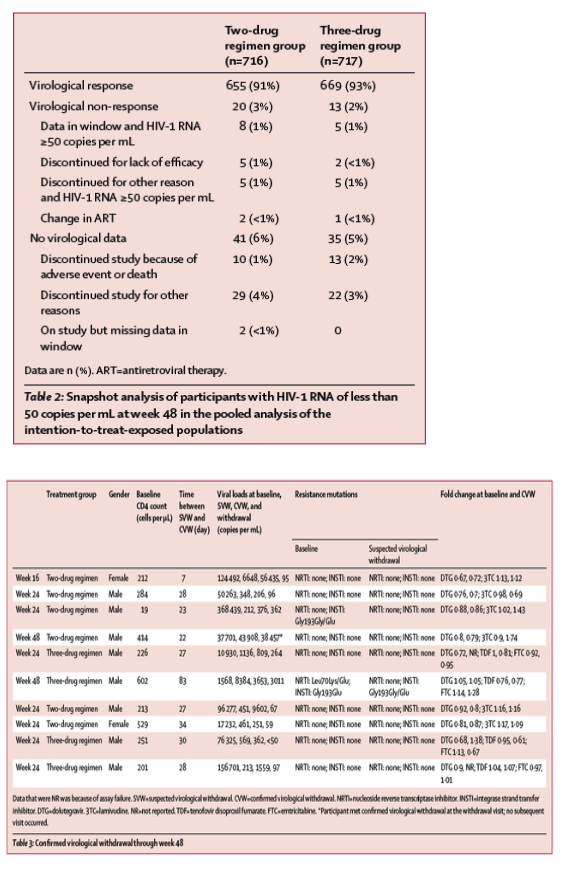
In the GEMINI-1 intention-to-treat-exposed population, 320 (90%) of 356 participants in the two-drug regimen group and 332 (93%) of 358 in the three-drug regimen group achieved HIV-1 RNA of less than 50 copies per mL at week 48 (adjusted treatment difference -2⋅6%, 95% CI -6⋅7 to 1⋅5; figure 2A); the corresponding number of participants in GEMINI-2 were 335 (93%) of 360 in the two-drug regimen group and 337 (94%) of 359 in the three-drug regimen group (adjusted treatment difference -0⋅7%, 95% CI -4⋅3 to 2⋅9; figure 2A). Each study met its primary efficacy endpoint of non-inferior virological efficacy at week 48, as the lower boundary of the two-sided 95% CI for the difference was greater than -10%. In the pooled analysis, 655 (91%) of 716 participants in the two-drug regimen group and 669 (93%) of 717 in the three-drug regimen group had HIV-1 RNA of less than 50 copies per mL at week 48 (adjusted treatment difference -1⋅7%, 95% CI -4⋅4 to 1⋅1; figure 2B). Virological non-response (defined by Snapshot analysis as ≥50 copies per mL at week 48) was observed in 20 (3%) of 716 participants in the two-drug regimen group versus 13 (2%) of 717 in the three-drug regimen group (table 2).
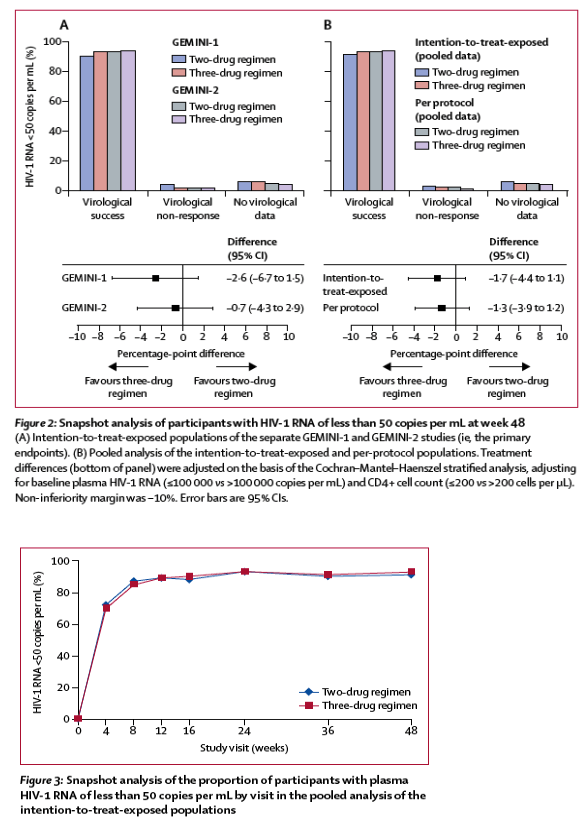

In the GEMINI-1 per-protocol population, 317 (92%) of 345 participants in the two-drug regimen group and 325 (94%) of 346 in the three-drug regimen group achieved HIV-1 RNA of less than 50 copies per mL at week 48 (adjusted treatment difference -1⋅9%, 95% CI -5⋅7 to 1⋅9); the corresponding number of participants in GEMINI-2 was 328 (94%) of 349 in the two-drug regimen group and 329 (95%) of 347 in the three-drug regimen group (adjusted treatment difference -0⋅7%, 95% CI -4⋅1 to 2⋅7). In the pooled per-protocol population, 645 (93%) of 694 participants in the two-drug regimen group versus 654 (94%) of 693 in the three-drug regimen group achieved HIV-1 RNA of less than 50 copies per mL (adjusted treatment difference -1⋅3%, 95% CI -3⋅9 to 1⋅2; figure 2B). The proportion of participants who achieved a response was high and similar between both treatment groups at all visits, and most participants achieved a plasma HIV-1 RNA of less than 50 copies per mL by week 4 (figure 3). A rapid decline of viral load was observed (appendix p 6), with a median time to viral suppression of 29⋅0 days in both treatment groups (IQR 29⋅0-55⋅0 for the two-drug regimen group and 29⋅0-57⋅0 days for the three-drug regimen group).
Ten (<1%) participants overall (six in the two-drug regimen group and four in the three-drug regimen group) met prespecified criteria for confirmed virological withdrawal through week 48. Genotypic testing of the HIV-1 reverse transcriptase, protease-reverse transcriptase, and integrase (IN) genes was successful for baseline and virological withdrawal samples from all ten participants, except for one IN genotype assay failure for one participant in the three-drug regimen group. For those with successfully amplified and sequenced samples, none had emergence of mutations conferring resistance to integrase strand transfer inhibitors or nucleoside reverse transcriptase inhibitors (table 3). All ten participants meeting confirmed virological withdrawal criteria were classified as virological rebounds.
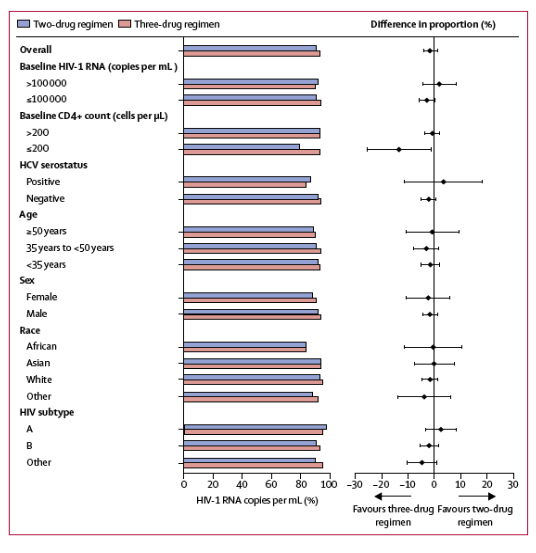
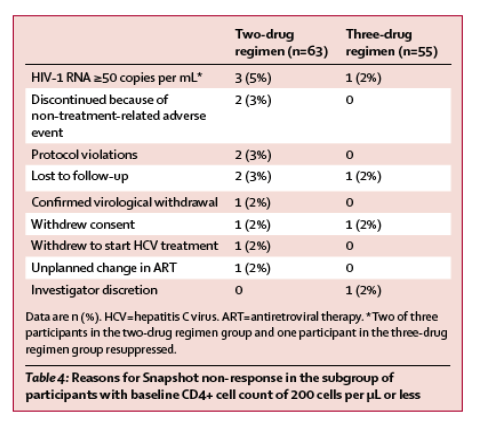
Response rates analysed by demographic and baseline characteristics were generally consistent with the overall response (figure 4). In the pooled analysis of the intention-to-treat-exposed population, 129 (92%) of 140 participants in the two-drug regimen group with baseline HIV-1 RNA of more than 100 000 copies per mL achieved virological success compared with 138 (90%) of 153 in the three-drug regimen group; 2% of participants in both groups had baseline viral loads of 500 000 copies per mL or more (after screening viral load <500 000 copies per mL). Efficacy outcomes in these participants were similar in the two-drug regimen group (11 [85%] of 13) and three-drug regimen group (12 [80%] of 15). In participants with baseline HIV-1 RNA of 100 000 copies per mL or less, 526 [91%] of 576 in the two-drug regimen and 531 [94%] of 564 in the three-drug regimen achieved virological success. In participants with CD4+ counts of more than 200 cells per μL, 605 (93%) of 653 in the two-drug regimen and 618 (93%) of 662 in the three-drug regimen achieved virological success. In participants with CD4+ counts of 200 cells per μL or less, 50 (79%) of 63 participants in the two-drug regimen group achieved HIV-1 RNA values of less than 50 copies per mL compared with 51 (93%) of 55 in the three-drug regimen group. Most reasons for Snapshot failures (ie, participants who did not achieve HIV-1 RNA <50 copies per mL at week 48) for this subgroup were unrelated to efficacy or treatment failure (table 4).
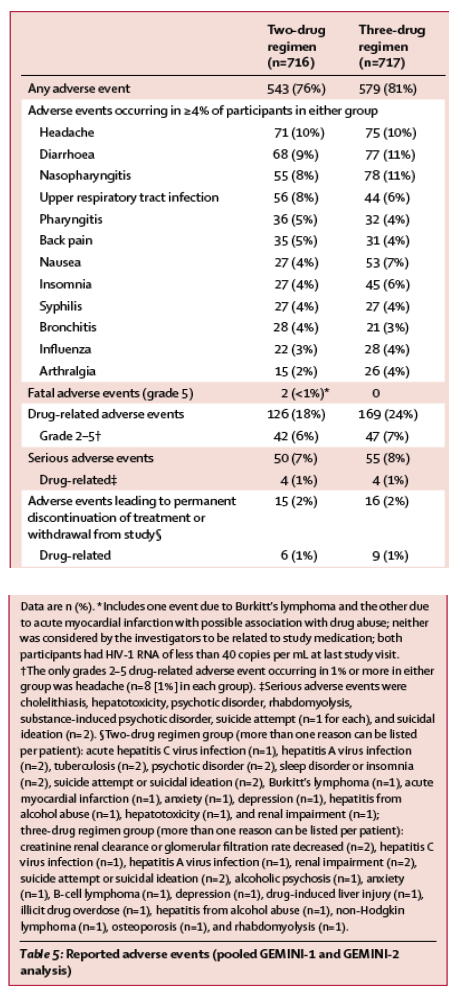
The most commonly reported adverse events across both treatment groups were headache, diarrhoea, nasopharyngitis, upper respiratory tract infection, nausea, and insomnia (table 5). Numerically, fewer participants reported drug-related adverse events in the two-drug regimen group than in the three-drug regimen group (126 [18%] of 716 vs 169 [24%] of 717). Differences in drug-related adverse events were primarily explained by lower frequencies of drug-related grade 1 nausea in the two-drug regimen group than in the three-drug regimen group (12 [2%] vs 33 [5%]). In the safety analysis, similar numbers of participants in the two-drug regimen group (15 [2%]) and three-drug regimen group (16 [2%]) had adverse events that led to permanent discontinuation of study drug. Similar proportions of serious adverse events were observed in the two-drug regimen group (50 [7%]) and three-drug regimen group (55 [8%]), with no single disorder reported as a serious adverse event in more than 1% of participants in either group. Two deaths were reported, both in the two-drug regimen group of GEMINI-2: one due to Burkitt's lymphoma and the other due to acute myocardial infarction with possible association with drug abuse. Neither death was considered by the investigators to be related to the study medication. The adverse event profile was similar between studies (appendix p 5).
Overall adverse events related to suicidal ideation and behaviour occurred in 29 participants: 17 (2%) in the two-drug regimen group (nine had suicidal ideation, three had suicidal behaviour, and five attempted suicide) and 12 (2%) in the three-drug regimen group (nine had suicidal ideation, one had suicidal behaviour, one attempted suicide, and one had intentional drug overdose). 13 (76%) of 17 in the two-drug regimen group and seven (58%) of 12 in the three-drug regimen group had a previous history of depression or suicidal behaviour or other psychiatric events.
Two pregnancies resulted in spontaneous abortions at 7 weeks and 4-5 weeks of gestation, and two resulted in elective abortions both at 6 weeks of gestation. No apparent congenital abnormalities were detected.
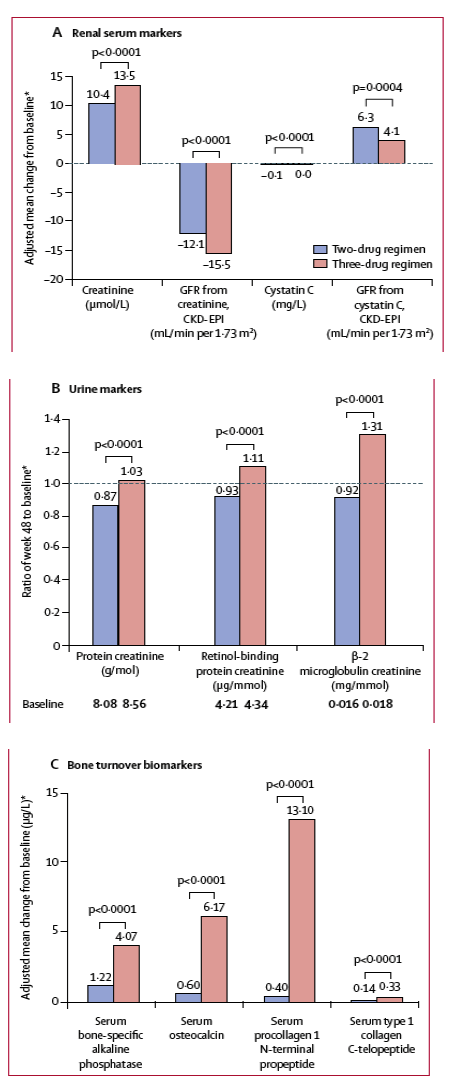
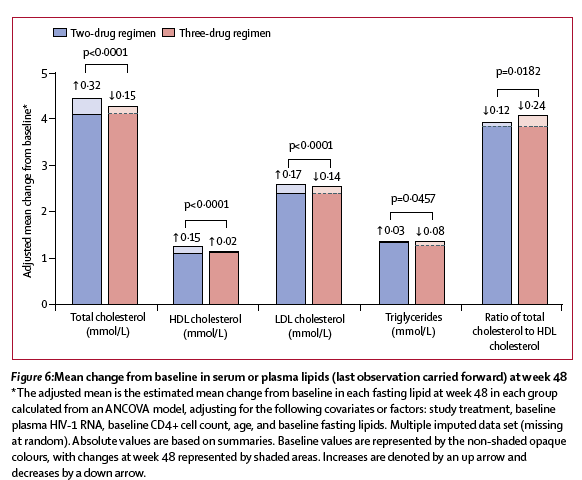
Changes in renal biomarkers were generally favourable in the two-drug regimen group compared with the three-drug regimen group (figures 5A, 5B). Increases in bone turnover biomarkers were observed in both treatment groups at week 48 (figure 5C); the magnitudes of the increases were smaller in the two-drug regimen group than in the three-drug regimen group. Changes in lipid parameters at week 48 varied between parameters and treatment groups (figure 6). Total cholesterol, LDL cholesterol, and total triglycerides increased from baseline to week 48 in the two-drug regimen group and decreased in the three-drug regimen group, with the differences between groups being significant for each. A significantly greater increase was observed in HDL cholesterol in the two-drug regimen group than in the three-drug regimen group. Small decreases in total cholesterol-to-HDL cholesterol ratio were observed, but the decrease in the three-drug regimen group was significantly greater than in the two-drug regimen group.
Mean baseline scores were high in both treatment groups on the EQ-5D-5L health state utility scale (0⋅950 [SD 0⋅076] for 716 participants in the two-drug regimen group; 0⋅940 [0⋅105] for 716 in the three-drug regimen group) and VAS (86⋅5 SD [12⋅1] for 715 in the two-drug regimen group; 85⋅1 [13⋅4] for 714 in the three-drug regimen group). At week 48, mean scores remained high for the health state utility scale (0⋅963 [SD 0⋅078] for 712 participants in the two-drug regimen group; 0⋅961 [0⋅076] for 710 in the three-drug regimen group) and VAS (90⋅2 [SD 10⋅0] for 712 in the two-drug regimen group; 89⋅3 [10⋅5] for 710 in the three-drug regimen group). For both scales, the between-group differences in the adjusted mean change from baseline to week 48 were not significant (health state utility score -0⋅0008 [95% CI -0⋅0078 to 0⋅0063]; p=0⋅834; and VAS score 0⋅4 [95% CI -0⋅6 to 1⋅3]; p=0⋅432).
|
|
| |
| |
|
|
|