 |
|
|
| |
Epigenetic Age Acceleration and Non-AIDS Defining
Cancers Among HIV Infected Adults
|
| |
| |
10th International Workshop on HIV and Aging, October 10-11, 2019, New York
Reported by Jules Levin
Chad J. Achenbach1, Yinan Zheng1, Brian Joyce1, Jun Wang1, Drew R. Nannini1, Jeffrey N. Martin2, W.C. Matthews3, Richard D. Moore4, Benigno Rodriguez5, Kenneth H. Meyer6, Joseph Eron7, Mari Kitahata8, Michael Saag9, and Lifang Hou1, on behalf of the CFAR Network of Integrated Clinical Systems (CNICS); 1Northwestern University, Chicago, IL, USA; 2University of California San Francisco, San Francisco, CA, USA; 3University of California San Diego, San Diego, CA, USA; 4Johns Hopkins University, Baltimore, MD, USA;5Case Western Reserve University, Cleveland, OH, USA;6Fenway Health, Boston, MA, USA;7University of North Carolina at Chapel Hill, Chapel Hill, NC, USA8University of Alabama at Birmingham, Birmingham, AL, USA;9University of Washington, Seattle, WA, USA

In conclusion, in these carefully matched small case-control studies, we found that only GrimAge acceleration was associated with lung cancer among individuals with HIV on ART and we believe this suggests that current epigenetic age measures may not perform the same or consistently in HIV infected individuals. Also that there are differences in magnitude of associations for specific age-related diseases including individual non-AIDS cancer types.
GrimAge captures epigenetic markers associated with oxidative stress and smoking and our findings may suggest that accumulation of molecular immunologic damage could be associated with lung cancer in persons with HIV independent of immune suppression measured by CD4 cell count. Our suspicion is that the association we observed here is largely due to residual confounding on extent of smoking that is captured better by methylation of CpG sites and DNA methylation patterns than clinical data available on smoking in CNICS. The causal role of DNA methylation patterns in the development of lung cancer is unclear, but GrimAge acceleration could be an important predictive biomarker for those aging with HIV.

These were pilot studies and we have further work we need to do in this area including studying epigenetic age acceleration among HIV populations and evaluating associations with other age related co-morbidities including cancer, cardiovascular disease, frailty and neurocognitive disorders in larger HIV cohorts
We also need to determine whether we are assessing epigenetic age in the best cell types or whether this should be measured in specific immune cell phenotypes such as subsets of CD8+ T cells
Finally, we wonder whether it would be useful to create a novel epigenetic age acceleration measure based on patterns of DNA methylation that correlate with inflammatory markers or immunologic changes specific to persons aging with HIV.

With potent ART classic AIDS defining cancers such as Kaposi sarcoma and central nervous system (CNS) lymphoma have declined; but non-AIDS cancers such as anal, lung and prostate cancer are an increasing cause of morbidity and mortality among our aging HIV populations.
Methylation of cytosine residues within CpG dinucleotides (5-methyl-cytosine) is one of several known epigenetic mechanisms and a vast literature characterizes CpG sites and/or genomic regions that become either hypermethylated or hypomethylated with increasing age, suggesting a role for DNA methylation in biological aging
Cross sectional studies have observed epigenetic age acceleration for those with HIV both on and off ART compared to uninfected individuals; however, we don't know whether epigenetic age acceleration is associated with age related diseases such as cancer among those with HIV or ultimately if these biomarkers could be used to predict disease in this population

To study this, we performed two match case-control pilot studies within 8 CNICS site across the U.S.
We identified cases as HIV infected adults with verified incident lung or anal cancer and PBMCs banked at least 6 months prior to the cancer event. We then selected controls by incidence density sampling from the CNICS primary study base as HIV infected adults without lung or anal cancer in care at the time of the index case diagnosis who were matched by age, sex, smoking status, and availability of a banked PBMC sample within 1 to 4 years prior to the index cancer case.
Epigenomic profiling was performed on DNA extracted from total PBMCs using the Illumina MethylationEPIC Beadchip (~850,000 sites). From these results, we calculated four measures of epigenetic age acceleration that I will describe in more detail on the next slide. We then performed conditional logistic regression, adjusting for race and CD4 count, to assess the associations between epigenetic age acceleration measures and incident cancer.

Since we may not all be familiar with epigenetic clocks I thought I would explain each of these measures a bit further.
Generally, the epigenetic clock was developed as an age predictor and deviations of DNAm Age from chronological age is of biological and potentially clinical interest. The idea here is that when DNAm Age exceeds chronological age, then the person is experiencing epigenetic age acceleration. This is defined as the residual from regressing DNAmAge on chronological age where a positive value indicates that epigenetic age is greater than expected.
This is then further characterized in blood as either intrinsic or extrinsic. Intrinsic refers to the residual from regressing the Horvath estimate of epigenetic age on chronological age and measures of blood cell counts. Intrinsic EAA accounts for age-related changes in blood cell compositions and captures aspects of epigenetic aging preserved across cell and tissue types.
Extrinsic Epigenetic Age Acceleration (EEAA) is derived from the Hannum estimate of epigenetic age and we do this by upweighting blood cell composition before regressing on chronological age. By increasing contribution of certain blood cell types that are changing with age, EEAA is able to capture aspects of immunosenescnece.
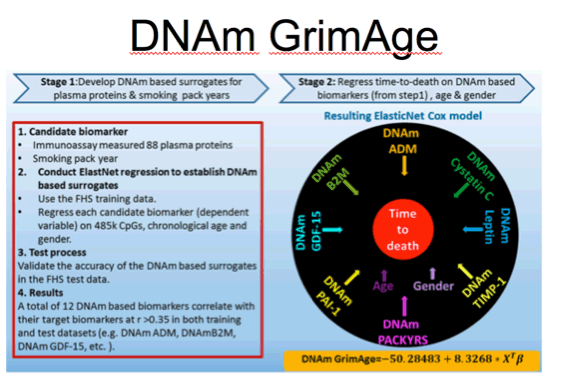
Lu et al. Aging, 2019 vol. 11 (2), pp. 303-327
This is a figure illustrating how the GrimAge measure was derived. Focus on this black circle and you can see the DNAm based biomarkers that go into this estimate including those that correlate with 7 plasma protein levels including beta-2 microglobulin, cystatin C, leptin, and plasminogen activation inhibitor 1.
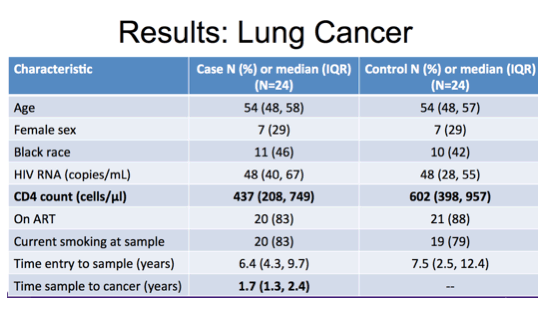
For the lung cancer study we included 24 cases and 24 matched controls as described in this table. You can see they were of similar age, sex and racial demographic distribution. The majority of both cases and controls were on ART with suppressed viral load at the time of sampling and similar high proportion of current smokers. The most notable difference between these groups was in median CD4 cell count with 437 cells in the cases compared to 602 in controls.
Of note, the timing of sample prior to cancer event was a median of nearly 2 years.
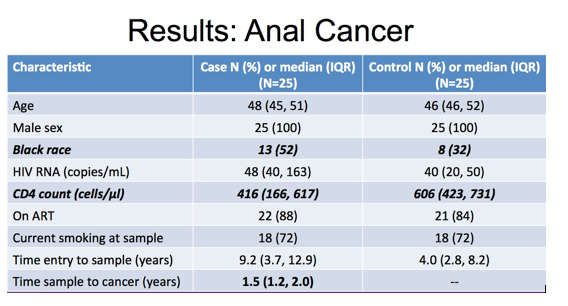
And here is the table for the anal cancer study that included 25 cases and 25 controls. Again, they were well matched on age, all were male, and similar high levels of current smoking. Again, the majority were on ART with suppressed viral load and notable differences were in proportion black race and median CD4 cell count at time of PBMC sample of 416 in cases and 606 in controls. The time of sample prior to anal cancer event was a median of one and a half years.

This table describes our primary study results and main findings of associations between 5-year epigenetic age acceleration measures and these two types of cancer. First, you can see that we did not find any significant associations between these epigenetic age measures and anal cancer. For lung cancer only GrimAge Acceleration was significantly associated with 316% greater odds per 5-year increase in age acceleration.

First and foremost I would like to thank the CNICS participants who contributed their time, data and samples to this study. Also my colleagues in the Northwestern Center for Population Epigenetics who performed these assays and the epigenetics analytics. We received funding through a Pilot Grant on Non-AIDS Cancers from the Lurie Comprehensive Cancer Center and Third Coast Center for AIDS Research. The TC CFAR also provided me funding to attend this conference and present our findings. Thank you and I am happy to take any questions.
|
| |
|
|
|
|
|