| |
Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK
|
| |
| |
Download the PDF here
Dec 8 2020 Lancet
In this interim analysis, we have not been able to assess duration of protection, since the first trials were initiated in April, 2020, such that all disease episodes have accrued within 6 months of the first dose being administered. Further evidence will be required to determine duration of protection and the need for additional booster doses of vaccine.
The results presented in this Article constitute the key findings from the first interim analysis, which are provided for rapid review by the public and policy makers. In future analyses with additional data included as they accrue, we will investigate differences in key subgroups such as older cohorts, ethnicity, dose regimen, and timing of booster vaccines, and we will search for correlates of protection.
Efficacy of 90⋅0% seen in those who received a low dose as prime in the UK was intriguingly high compared with the other findings in the study. Although there is a possibility that chance might play a part in such divergent results, a similar contrast in efficacy between the LD/SD and SD/SD recipients with asymptomatic infections provides support for the observation (58⋅9% [95% CI 1⋅0 to 82⋅9] vs 3⋅8% [-72⋅4 to 46⋅3]). Exploratory subgroup analyses, included at the request of reviewers and editors, that were restricted to participants aged 18-55 years, or aligned (>8 weeks) intervals between doses, showed similar findings. Use of a low dose for priming could provide substantially more vaccine for distribution at a time of constrained supply, and these data imply that this would not compromise protection. While a vaccine that could prevent COVID-19 would have a substantial public health benefit, prevention of asymptomatic infection could reduce viral transmission and protect those with underlying health conditions who do not respond to vaccination, those who cannot be vaccinated for health reasons, and those who will not or cannot access a vaccine, providing wider benefit for society. However, the wide CIs around our estimates show that further data are needed to confirm these preliminary findings, which will be done in future analyses of the data accruing in these ongoing trials. Similar results have been seen for other vaccines where a reduced number or type of priming dose in infancy can lead to higher responses to a booster vaccine. Further work is needed to determine the mechanism of the increased efficacy with a LD/SD regimen, which might be due to higher levels of neutralising antibody, lower levels of anti-vector immunity with lower vector-derived antigen content of the first dose, or differential antibody functionality or cellular immunity, including altered avidity or immunodominance.
Other coronavirus vaccine developers have released preliminary high-level results in public statements, including more than 90% efficacy reported for the lipid nanoparticle mRNA vaccine BNT162b2, 92% efficacy for the Sputnik V vaccine (developed at the National Research Centre for Epidemiology and Microbiology), and 94⋅5% for the Moderna lipid nanoparticle mRNA-1273 vaccine.
The possibility that more than one efficacious vaccine against COVID-19 might be approved for use in the near future is encouraging. However, control of pandemic coronavirus will only be achieved if the licensure, manufacturing, and distribution of these vaccines can be achieved at an unprecedented scale and vaccination is rolled out to all those who are vulnerable.
While the data presented here show that ChAdOx1 nCov-19 is efficacious against symptomatic disease, with most cases accruing in adults younger than 55 years of age so far, an important public health consideration is the morbidity and mortality of the disease in an older adult population and thus the potential efficacy in this age group. We have reported immunogenicity data showing similar immune responses following vaccination with two doses of ChAdOx1 nCov-19 in older adults, including those older than 70 years of age, when compared with those younger than 55 years.
https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(20)32661-1/fulltext
Dec 8 2020
Summary
The primary objective was to evaluate the efficacy of ChAdOx1 nCoV-19 vaccine against NAAT-confirmed COVID-19. The primary outcome was virologically confirmed, symptomatic COVID-19, defined as a NAAT-positive swab combined with at least one qualifying symptom (fever ≥37⋅8°C, cough, shortness of breath, or anosmia or ageusia).
Participants in the ChAdOx1 nCoV-19 group received two doses containing 5 × 1010 viral particles (standard dose; SD/SD cohort); a subset in the UK trial received a half dose as their first dose (low dose) and a standard dose as their second dose (LD/SD cohort).
Background
A safe and efficacious vaccine against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), if deployed with high coverage, could contribute to the control of the COVID-19 pandemic. We evaluated the safety and efficacy of the ChAdOx1 nCoV-19 vaccine in a pooled interim analysis of four trials.
Baseline assessments included review of inclusion and exclusion criteria, medical history, vital signs measurement, history-directed clinical examination, and collection of serum for SARS-CoV-2 serology. To test for asymptomatic infections, participants in COV002 in the UK were asked to provide a weekly self-administered nose and throat swab for NAAT testing from 1 week after first vaccination using kits provided by the UK Department of Health and Social Care (DHSC). Participants were given home test kits provided by the DHSC that included step-by-step instructions on how to do a self-swab and a link to a demonstration video. The site trial team provided support with logistics of packaging and returning test kits and tracking swab results to participants if required. Swabs were taken by participants in their homes and posted to dedicated DHSC testing laboratories for processing. Participants were directly informed of their results by text or email from the National Health Service (NHS). Swab results from participants in England and Wales were provided to the trial statistician on a daily basis by the NHS and matched to individuals based on personal identification data (name, date of birth, NHS number, and postcode). Swab results from participants in Scotland were unavailable to the study team at the time of the data cutoff for this analysis, but will be included in future analyses. Any swab results that were not able to be matched to a study participant using at least two pieces of personal data were not added to the study database.
For symptomatic participants in COV002 in the UK, weekly swabbing continued both before and after participants reported symptoms to the study site. Thus, a participant who reported symptoms and was clinically assessed might also have had additional swabs return positive results through the asymptomatic testing process for several weeks. In addition, due to the large number of health-care workers enrolled in these studies, some participants were tested according to their workplace testing policies and these results were also entered into the database for review by the masked endpoint evaluation committee. Further exploratory assessment of the length of time participants remained NAAT-positive, and the sources of information used for case detection will be done in future analyses.
Methods
This analysis includes data from four ongoing blinded, randomised, controlled trials done across the UK, Brazil, and South Africa. Three of the studies are single blind and one is double blind (COV005).
Participants aged 18 years and older were randomly assigned (1:1) to ChAdOx1 nCoV-19 vaccine or control (meningococcal group A, C, W, and Y conjugate vaccine or saline). Participants in the ChAdOx1 nCoV-19 group received two doses containing 5 × 1010 viral particles (standard dose; SD/SD cohort); a subset in the UK trial received a half dose as their first dose (low dose) and a standard dose as their second dose (LD/SD cohort). The primary efficacy analysis included symptomatic COVID-19 in seronegative participants with a nucleic acid amplification test-positive swab more than 14 days after a second dose of vaccine. Participants were analysed according to treatment received, with data cutoff on Nov 4, 2020. Vaccine efficacy was calculated as 1 - relative risk derived from a robust Poisson regression model adjusted for age. Studies are registered at ISRCTN89951424 and ClinicalTrials.gov, NCT04324606, NCT04400838, and NCT04444674.
Findings
Between April 23 and Nov 4, 2020, 23 848 participants were enrolled and 11 636 participants (7548 in the UK, 4088 in Brazil) were included in the interim primary efficacy analysis. In participants who received two standard doses, vaccine efficacy was 62⋅1% (95% CI 41⋅0-75⋅7; 27 [0⋅6%] of 4440 in the ChAdOx1 nCoV-19 group vs71 [1⋅6%] of 4455 in the control group) and in participants who received a low dose followed by a standard dose, efficacy was 90⋅0% (67⋅4-97⋅0; three [0⋅2%] of 1367 vs 30 [2⋅2%] of 1374; pinteraction=0⋅010). Overall vaccine efficacy across both groups was 70⋅4% (95⋅8% CI 54⋅8-80⋅6; 30 [0⋅5%] of 5807 vs 101 [1⋅7%] of 5829). From 21 days after the first dose, there were ten cases hospitalised for COVID-19, all in the control arm; two were classified as severe COVID-19, including one death. There were 74 341 person-months of safety follow-up (median 3⋅4 months, IQR 1⋅3-4⋅8): 175 severe adverse events occurred in 168 participants, 84 events in the ChAdOx1 nCoV-19 group and 91 in the control group. Three events were classified as possibly related to a vaccine: one in the ChAdOx1 nCoV-19 group, one in the control group, and one in a participant who remains masked to group allocation.
Five cases included in the primary analysis occurred in those participants older than 55 years of age. Vaccine efficacy in older age groups could not be assessed but will be determined, if sufficient data are available, in a future analysis after more cases have accrued.
Interpretation
ChAdOx1 nCoV-19 has an acceptable safety profile and has been found to be efficacious against symptomatic COVID-19 in this interim analysis of ongoing clinical trials.
COV001 (UK)
COV001 is a continuing single-blind phase 1/2 clinical trial in five sites in the UK, which began on April 23, 2020, and enrolled 1077 healthy volunteers aged 18-55 years, as previously described.
Briefly, healthy adult participants were enrolled after screening to exclude those with pre-existing health conditions. Participants were randomly assigned 1:1 to receive ChAdOx1 nCoV-19 at a dose of 5 × 1010 viral particles (standard dose), measured using spectrophotometry, or meningococcal group A, C, W, and Y conjugate vaccine (MenACWY) as control. An open-label non-randomised subgroup of ten participants were given two doses of ChAdOx1 nCoV-19 28 days apart, as previously reported.
This study was originally planned as a single-dose study and 88 participants in the phase 1 part of the study remain recipients of a single dose. However, the protocol was modified to a two-dose regime, following an amendment on July 30, 2020 (version 9.0; appendix 2 pp 180-181), for the remaining phase 2 cohorts as a result of robust booster responses identified in the evaluation of the early immunogenicity cohorts, with the booster dose given at the earliest possible time.
COV002 (UK)
COV002 is a continuing single-blind phase 2/3 study in the UK that began on May 28, 2020, and enrolled participants in 19 study sites in England, Wales, and Scotland. Enrolment particularly targeted individuals working in professions with high possible exposure to SARS-CoV-2, such as health and social care settings.
Two dosage groups were included in COV002: participants who received a low dose of the vaccine (2⋅2 × 1010 viral particles) as their first dose and were boosted with a standard dose (in the LD/SD group), and subsequent cohorts who were vaccinated with two standard-dose vaccines (SD/SD group).
COV003 (Brazil)
COV003 is a continuing single-blind phase 3 study in Brazil that began on June 23, 2020. The focus of recruitment was targeted at those at high risk of exposure to the virus, including health-care workers at six sites across Brazil. Participants were aged 18 years or older, and this trial included individuals with stable pre-existing health conditions. All participants were offered two doses of the vaccine at a dose of 3⋅5-6⋅5 × 1010 viral particles with administration up to 12 weeks apart (target 4 weeks), following a protocol amendment on July 28, 2020, to include booster groups (version 4.0; appendix 2 pp 438-439). Full details are available in the study protocol (appendix 2 pp 343-441).
COV005 (South Africa)
COV005 is a continuing double-blind phase 1/2 study in South Africa in healthy adults aged 18-65 years living without HIV that began on June 28, 2020. An additional immunogenicity cohort of those living with HIV was also enrolled but are not included in this interim analysis. All participants were offered two doses of the vaccine at a dose of 3⋅5-6⋅5 × 1010 viral particles, with doses administered 4 weeks apart. A small subgroup of 44 participants received a half-dose vaccine (21 as their first dose and 23 as their second dose) as a result of variability in the release assay, before the adoption of new methods for characterisation of concentration. Adjustment in dose was discussed with and approved by the national regulator. Full details are available in the study protocol (appendix 2 pp 442-559).
Results
Between April 23 and Nov 4, 2020, 23 848 participants were recruited and vaccinated across the four studies: 1077 in COV001 (UK), 10 673 in COV002 (UK), 10 002 in COV003 (Brazil), and 2096 in COV005 (South Africa). 11 636 participants in COV002 and COV003 met the inclusion criteria for the primary analysis, 5807 of whom received two doses of ChAdOx1 nCoV-19 and 5829 of whom received two doses of control product. A trial profile and reasons for exclusion from the primary analysis are shown in appendix 1 (pp 5-7). Here, we provide safety data on 74 341 person-months of follow-up after first dose (median 3⋅4 months, IQR 1⋅3-4⋅8) and 29 060 person-months of follow-up after two doses (median 2⋅0, 1⋅3-2⋅3).
Of the participants in COV002 and COV003 included in the primary efficacy analyses, the majority were aged 18-55 years (6542 [86⋅7%] of 7548 in the UK and 3676 [89⋅9%] of 4088 in Brazil; table 1). Those aged 56 years or older were recruited later and contributed 12⋅2% of the total cohort in the current analysis (1006 [13⋅3%] in the UK and 412 [10⋅1%] in Brazil). 7045 (60⋅5%) participants were female. 6902 (91⋅4%) participants in the UK and 2723 (66⋅6%) participants in Brazil were white (table 1). Baseline participants of the safety population are shown in appendix 1 (pp 9-10).
The timing of priming and booster vaccine administration varied between studies. As protocol amendments to add a booster dose took place when the trials were underway, and owing to the time taken to manufacture and release a new batch of vaccine, doses could not be administered at a 4-week interval. 1459 (53⋅2%) of 2741 participants in COV002 in the LD/SD group received a second dose at least 12 weeks after the first (median 84 days, IQR 77—91) and only 22 (0⋅8%) received a second dose within 8 weeks of the first. The median interval between doses for the SD/SD group in COV002 was 69 days (50-86). Conversely, the majority of participants in COV003 in the SD/SD group (2493 [61⋅0%] of 4088) received a second dose within 6 weeks of the first (median 36 days, 32-58; appendix 1 p 11).
A small proportion of participants were seropositive at baseline (138 [1⋅3%] of 10 673 in the UK and 235 [2⋅3%] of 10 002 in Brazil). Three participants seropositive at baseline had subsequent NAAT-positive swabs. One participant had an asymptomatic infection 3 weeks after a first dose of ChAdOx1 nCoV-19. Two other participants in the control group had symptomatic infections 8 weeks and 21 weeks after their baseline sample was taken.
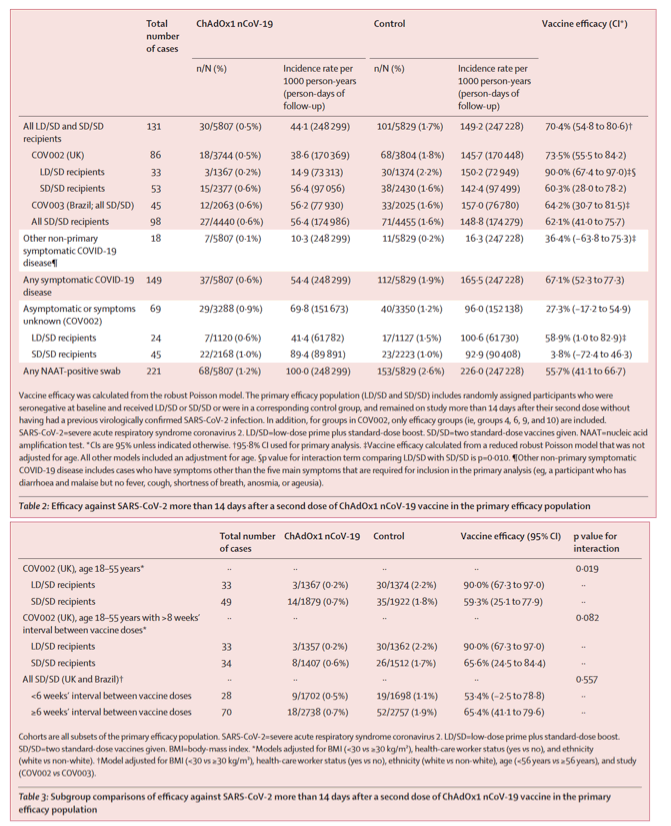
There were 131 cases of symptomatic COVID-19 in LD/SD or SD/SD recipients who were eligible for inclusion in the primary efficacy analysis more than 14 days after the second dose of vaccine (table 2). There were 30 (0⋅5%) cases among 5807 participants in the vaccine arm and 101 (1⋅7%) cases among 5829 participants in the control group, resulting in vaccine efficacy of 70⋅4% (95⋅8% CI 54⋅8-80⋅6; table 2; figure). In participants who received two standard-dose vaccines, vaccine efficacy was 62⋅1% (95% CI 41⋅0-75⋅7), whereas in those who received a low dose as their first dose of vaccine, efficacy was higher at 90⋅0% (67⋅4-97⋅0; pinteraction=0⋅010; table 2; appendix 1 pp 12-13).
In England and Wales, 129 529 weekly self-swabs were processed by the DHSC, of which 126 324 (97⋅5%) were matched to study participants. There were 435 positive swabs, of which 354 (81⋅4%) were matched. Symptoms in these participants were not routinely assessed as swabs were done at home and sent for testing through the post. Asymptomatic infections or those with unreported symptoms were detected in 69 participants (table 2). Vaccine efficacy in the 24 LD/SD recipients was 58⋅9% (95% CI 1⋅0 to 82⋅9), whereas it was 3⋅8% (-72⋅4 to 46⋅3) in the 45 participants receiving SD/SD (table 2).
Results from the subgroup comparisons presented in this analysis were similar to overall results (table 3). In the SD/SD UK cohort who were aged 18-55 years, 49 cases were available for inclusion in the analysis and vaccine efficacy was 59⋅3% (95% CI 25⋅1 to 77⋅9; pinteraction=0⋅019; table 3). When further restricted to those who received their vaccines more than 8 weeks apart, 33 cases were included in the SD/SD analysis and vaccine efficacy was 65⋅6% (24⋅5 to 84⋅4; pinteraction=0⋅082; table 3; appendix 1 pp 12-13). In the SD/SD cohorts in the UK and Brazil, vaccine efficacy was similar when analysed in subgroups according to time between vaccines, at 53⋅4% (-2⋅5 to 78⋅8) in participants with less than 6 weeks’ interval between doses and 65⋅4% (41⋅1 to 79⋅6) in participants with at least 6 weeks’ interval (pinteraction=0⋅56; table 3).
For our secondary analysis of cases occurring more than 21 days after the first standard dose in participants who received only standard doses, there were 192 included cases with a vaccine efficacy of 64⋅1% (95% CI 50⋅5-73⋅9; table 4; figure)
Vaccine efficacy was calculated from the robust Poisson model. The first-standard-dose efficacy population includes participants seronegative at baseline who received only standard dose vaccines or were in the corresponding control group, and remained on study 22 days after their first dose without having had a previous virologically confirmed SARS-CoV-2 infection. In addition, for groups in COV002, only efficacy groups (ie, groups 4, 6, 9, and 10) are included. SARS-CoV-2=severe acute respiratory syndrome coronavirus 2. NAAT=nucleic acid amplification test.
The safety population includes all randomisation participants who received at least one dose of vaccine. Severe COVID-19 (WHO score ≥6) is a subset of hospitalisations (WHO score ≥4). Cases were eligible for inclusion in efficacy if the first symptom or first NAAT-positive result was on or before the data cutoff date (Nov 4, 2020). Two cases appear in this table that do not appear in the table for serious adverse events in appendix 1 (pp 15-20) as the adverse event reporting date was after the data cutoff date. MenACWY=meningococcal group A, C, W, and Y conjugate vaccine. NAAT=nucleic acid amplification test.
Five cases included in the primary analysis occurred in those participants older than 55 years of age. Vaccine efficacy in older age groups could not be assessed but will be determined, if sufficient data are available, in a future analysis after more cases have accrued.
Across all four studies, the vaccine had a good safety profile with serious adverse events and adverse events of special interest balanced across the study arms. Serious adverse events occurred in 168 participants, 79 of whom received ChAdOx1 nCoV-19 and 89 of whom received MenACWY or saline control (appendix 1 pp 15-18). There were 175 events (84 in the ChAdOx1 nCoV-19 group and 91 in the control group), three of which were considered possibly related to either the experimental or a control vaccine. A case of haemolytic anaemia in the control group in the UK phase 1/2 study occurring 10 days after MenACWY vaccine was considered possibly related to the intervention and has been previously described.
A case of transverse myelitis was reported 14 days after ChAdOx1 nCoV-19 booster vaccination as being possibly related to vaccination, with the independent neurological committee considering the most likely diagnosis to be of an idiopathic, short segment, spinal cord demyelination. A potentially vaccine-related serious adverse event was reported 2 days after vaccination in South Africa in an individual who recorded fever higher than 40°C, but who recovered rapidly without an alternative diagnosis and was not admitted to hospital. The participant remains masked to group allocation, continues in the trial, and received a second dose of the allocated vaccine without a similar reaction.
There were two additional cases of transverse myelitis that were originally reported as potentially related but later determined to be unlikely to be related to vaccination by an independent committee of neurological experts. One case that occurred 10 days after a first vaccination with ChAdOx1 nCoV-19 was initially assessed as possibly related, but later considered unlikely to be related by the site investigator when further investigation revealed pre-existing, but previously unrecognised, multiple sclerosis. The second case was reported 68 days after MenACWY vaccination. While considered possibly related by the site investigator at the time of reporting, an independent panel of neurological experts considered this to be unlikely. All trial participants have recovered, or are in a stable or improving condition.
There were four non-COVID-19 deaths reported across the studies (three in the control arm and one in the ChAdOx1 nCoV-19 arm) that were all considered unrelated to the vaccine, with cause of death assessed as road traffic accident, blunt force trauma, homicide, and fungal pneumonia.
Discussion
Here, we present the first interim safety and efficacy data for a viral vector coronavirus vaccine, ChAdOx1 nCoV-19, evaluated in four trials across three continents, showing significant vaccine efficacy of 70⋅4% after two doses and protection of 64⋅1% after at least one standard dose, against symptomatic disease, with no safety concerns.
The prespecified analysis population, which was determined following feedback from national and international regulators before unblinding of the study, included a pooled analysis from several countries to improve generalisability, and inclusion of two dose subgroups within the UK trial. This pooling strategy was authorised by the chief investigator (AJP) and study statistician (MV), with no concerns about pooling different control groups, and was accepted by regulators involved in the discussions. There had been initial concern that the LD/SD regimen might have lower efficacy than SD/SD, and the regulatory authority acceptance of the inclusion of the two trial regimens (LD/SD and SD/SD) in analysis was based on the observation that these regimens generated similar levels of binding antibody, and would therefore increase the sample size available for analysis without compromising efficacy. The discussion about pooling and inclusion of LD/SD was made at a time when disease rates were low in the UK and, in the face of the pandemic, it was agreed that pooling could provide the earliest possible read on efficacy that could contribute to public health.
No previous trials have been published on the efficacy of a viral-vectored coronavirus vaccine and so this study provides the first peer-reviewed evidence that induction of immune responses against spike protein using viral vectors provides protection against the disease in humans, as has been seen in animal models.
In participants who received two standard doses, efficacy against primary symptomatic COVID-19 was consistent in both the UK (60⋅3% efficacy) and Brazil (64⋅2% efficacy), indicating these results are generalisable across two diverse settings with different timings for the booster dose (with most participants in the UK receiving the booster dose more than 12 weeks after the first dose and most participants in Brazil receiving their second dose within 6 weeks of the first). Exploratory subgroup analyses included at the request of reviewers and editors also showed no significant difference in efficacy estimates when comparing those with a short time window between doses (<6 weeks) and those with longer (≥6 weeks), although further detailed exploration of the timing of doses might be warranted.
Efficacy of 90⋅0% seen in those who received a low dose as prime in the UK was intriguingly high compared with the other findings in the study. Although there is a possibility that chance might play a part in such divergent results, a similar contrast in efficacy between the LD/SD and SD/SD recipients with asymptomatic infections provides support for the observation (58⋅9% [95% CI 1⋅0 to 82⋅9] vs 3⋅8% [-72⋅4 to 46⋅3]). Exploratory subgroup analyses, included at the request of reviewers and editors, that were restricted to participants aged 18-55 years, or aligned (>8 weeks) intervals between doses, showed similar findings. Use of a low dose for priming could provide substantially more vaccine for distribution at a time of constrained supply, and these data imply that this would not compromise protection. While a vaccine that could prevent COVID-19 would have a substantial public health benefit, prevention of asymptomatic infection could reduce viral transmission and protect those with underlying health conditions who do not respond to vaccination, those who cannot be vaccinated for health reasons, and those who will not or cannot access a vaccine, providing wider benefit for society. However, the wide CIs around our estimates show that further data are needed to confirm these preliminary findings, which will be done in future analyses of the data accruing in these ongoing trials.
Similar results have been seen for other vaccines where a reduced number or type of priming dose in infancy can lead to higher responses to a booster vaccine.
Further work is needed to determine the mechanism of the increased efficacy with a LD/SD regimen, which might be due to higher levels of neutralising antibody, lower levels of anti-vector immunity with lower vector-derived antigen content of the first dose, or differential antibody functionality or cellular immunity, including altered avidity or immunodominance.
Other coronavirus vaccine developers have released preliminary high-level results in public statements, including more than 90% efficacy reported for the lipid nanoparticle mRNA vaccine BNT162b2, 92% efficacy for the Sputnik V vaccine (developed at the National Research Centre for Epidemiology and Microbiology), and 94⋅5% for the Moderna lipid nanoparticle mRNA-1273 vaccine.
The possibility that more than one efficacious vaccine against COVID-19 might be approved for use in the near future is encouraging. However, control of pandemic coronavirus will only be achieved if the licensure, manufacturing, and distribution of these vaccines can be achieved at an unprecedented scale and vaccination is rolled out to all those who are vulnerable.
The US Food and Drug Administration's guidelines indicate that they would license a vaccine against the pandemic virus that showed at least 50% efficacy and WHO have indicated a minimum efficacy of 50% in its target product profile. A modelling study found that a vaccine with efficacy of 60-80% could allow reduction in physical distancing measures, but this would still require high coverage. The findings here indicate that the efficacy of ChAdOx1 nCoV-19 exceeds these thresholds and has the potential to have a public health impact.
Much consideration has been given to the statistical confidence in vaccine efficacy estimates, given the size of the global population who might be vaccinated. To ensure that point estimates of efficacy in clinical trials are sufficiently robust, some regulatory authorities consider that the lower bound of the CI for efficacy should be higher than 20% (personal communication), with other authorities more stringent and anticipating a lower bound of 30% for licensure.
Here, we present data that exceed both these thresholds in the pooled analysis, which we had agreed with regulators before unblinding of the study, and also meet the thresholds set in the individual analyses of trials by country and by study arm.
We designed our studies early in the pandemic and fixed our primary symptomatic disease endpoint on the basis of expert analysis and guidelines from Public Health England and WHO as the first wave of disease spread around the world, although these have now been substantially updated.
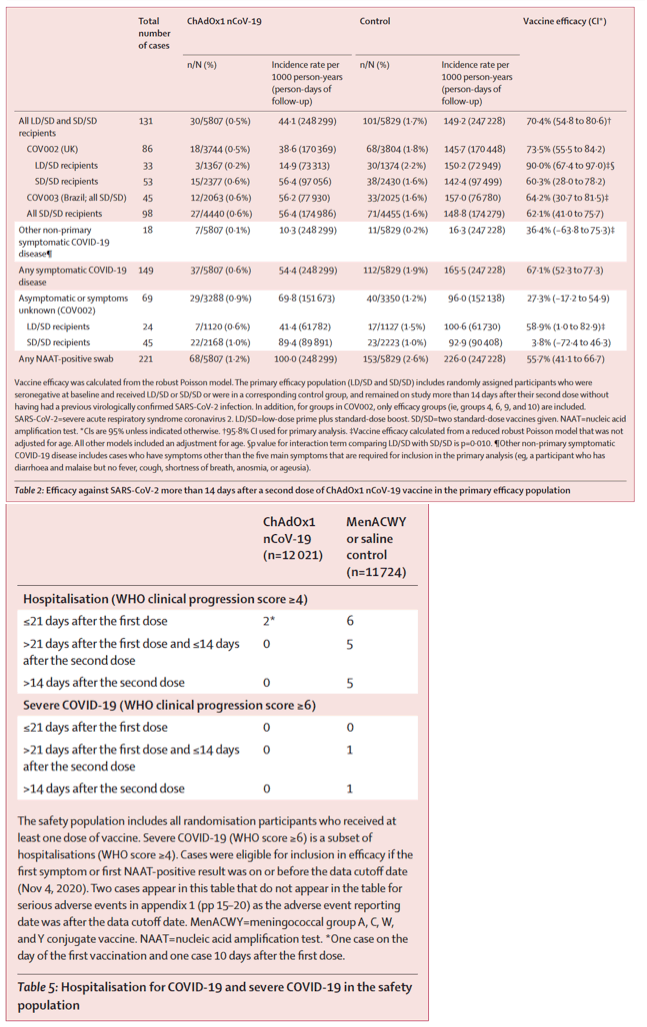
We have used a restricted definition of symptomatic disease, since many other symptoms that are associated with COVID-19 disease are non-specific. Since endpoints in protocols for different vaccines are not well aligned, we recognise that it will be difficult to compare efficacy across programmes. However, we have also included hospital admissions and severe disease as an endpoint in the current study, which might be easier to assess in comparison with other vaccines, and found that in the ten cases available for analysis more than 21 days after the first dose, there was complete protection against hospitalisation for COVID-19.
While the data presented here show that ChAdOx1 nCov-19 is efficacious against symptomatic disease, with most cases accruing in adults younger than 55 years of age so far, an important public health consideration is the morbidity and mortality of the disease in an older adult population and thus the potential efficacy in this age group. We have reported immunogenicity data showing similar immune responses following vaccination with two doses of ChAdOx1 nCov-19 in older adults, including those older than 70 years of age, when compared with those younger than 55 years.
As older age groups were recruited later than younger age groups, there has been less time for cases to accrue and as a result, efficacy data in these cohorts are currently limited by the small number of cases, but additional data will be available in future analyses.
These trials, conducted on three different continents, enrolled geographically and ethnically diverse populations. Severe COVID-19 has been seen to disproportionately affect people of non-white ethnicity, as well as those who are male, overweight, and the elderly.
In our studies, the demographic characteristics of those enrolled varied between countries. In the UK, the enrolled population was predominantly white and, in younger age groups, included more female participants due to the focus on enrolment of health-care workers. This is a typically lower risk population for severe COVID-19. The demographic profile combined with the weekly self-swabbing for asymptomatic infection in the UK results in a milder case-severity profile. In Brazil, there was a larger proportion of non-white ethnicities, and again the majority of those enrolled were health-care workers.
We have previously reported on the local and systemic reactogenicity of ChAdOx1 nCoV-19 and shown that it is tolerated and that the side-effects are less both in intensity and number in older adults, with lower doses, and after the second dose. Although there were many serious adverse events reported in the study in view of the size and health status of the population included, there was no pattern of these events that provided a safety signal in the study. Three cases of transverse myelitis were initially reported as suspected unexpected serious adverse reactions, with two in the ChAdOx1 nCoV-19 vaccine study arm, triggering a study pause for careful review in each case. Independent clinical review of these cases has indicated that one in the experimental group and one in the control group are unlikely to be related to study interventions, but a relationship remained possible in the third case. Careful monitoring of safety, including neurological events, continues in the trials. All safety data will be provided to regulators for review.
In this interim analysis, we have not been able to assess duration of protection, since the first trials were initiated in April, 2020, such that all disease episodes have accrued within 6 months of the first dose being administered. Further evidence will be required to determine duration of protection and the need for additional booster doses of vaccine.
The results presented in this Article constitute the key findings from the first interim analysis, which are provided for rapid review by the public and policy makers. In future analyses with additional data included as they accrue, we will investigate differences in key subgroups such as older cohorts, ethnicity, dose regimen, and timing of booster vaccines, and we will search for correlates of protection.
Until widespread immunity halts the spread of SARS-CoV-2, physical distancing measures and novel therapies are needed to control COVID-19. In the meantime, an efficacious vaccine has the potential to have a major impact on the pandemic if used in populations at risk of severe disease. Here, we have shown for the first time that a viral vector vaccine, ChAdOx1 nCoV-19, is efficacious and could contribute to control of the disease in this pandemic.
|
|
| |
| |
|
|
|