| |
|
Fluoxazolevir inhibits hepatitis C virus infection in humanized chimeric mice by blocking viral membrane fusion
|
| |
| |
Download the PDF here
Aug 2020 Nat Mirobiology - Christopher D. Ma 1, Michio Imamura2, Daniel C. Talley3, Adam Rolt1, Xin Xu3, Amy Q. Wang3,
Derek Le1, Takuro Uchida2, Mitsutaka Osawa2, Yuji Teraoka2, Kelin Li4, Xin Hu3, Seung Bum Park1,
Nishanth Chalasani1, Parker H. Irvin1, Andres E. Dulcey3, Noel Southall3, Juan J. Marugan3, Zongyi Hu1,
Kazuaki Chayama2, Kevin J. Frankowski 4 and Tsanyang Jake Liang 1
Fluoxazolevir is an aryloxazole-based entry inhibitor of hepatitis C virus (HCV).....Fluoxazolevir can achieve complete inhibition against all seven HCV chimeric genotypes in vitro with varying efficacies.......A 6-week combination therapy with fluoxazolevir and glecaprevir/pibrentasvir was conducted in humanized chimeric mice infected with this multidrug-resistant HCV strain......In combination-treated mice, HCV RNA decreased rapidly to undetectable levels and remained below detectable levels throughout the duration of treatment, indicating the effectiveness of fluoxazolevir together with glecaprevir/pibrentasvir in suppressing this multidrug-resistant variant.....These data suggest that an entry inhibitor, such as fluoxazolevir, can partially prevent and hinder the progression of de novo HCV infection in vivo...Efficacy studies in humanized Alb-uPA/Scid mice showed that fluoxazolevir significantly suppressed HCV RNA levels during either monotherapy or combination therapy with daclatasvir......A potent entry inhibitor, especially in combination with other DAAs, can minimize resistance and shorten treatment duration
.....Our preclinical studies support fluoxazolevir as a promising candidate for the next generation of drug cocktails for HCV treatment. It is synergistic with U.S. Food and Drug Administration-approved HCV antivirals, active against all HCV genotypes in vitro, has preferential localization in the liver, can clear various HCV strains in a humanized mouse model and has potential to delay or prevent acute HCV infection. Since the viral fusion structure and process is relatively conserved50, it is also tempting to speculate that fluoxazolevir may have broader antiviral activities against other viruses
Abstract
Fluoxazolevir is an aryloxazole-based entry inhibitor of hepatitis C virus (HCV). We show that fluoxazolevir inhibits fusion of HCV with hepatic cells by binding HCV envelope protein 1 to prevent fusion. Nine of ten fluoxazolevir resistance-associated substitutions are in envelope protein 1, and four are in a putative fusion peptide. Pharmacokinetic studies in mice, rats and dogs revealed that fluoxazolevir localizes to the liver. A 4-week intraperitoneal regimen of fluoxazolevir in humanized chimeric mice infected with HCV genotypes 1b, 2a or 3 resulted in a 2-log reduction in viraemia, without evidence of drug resistance. In comparison, daclatasvir, an approved HCV drug, suppressed more than 3 log of viraemia but is associated with the emergence of resistance-associated substitutions in mice. Combination therapy using fluoxazolevir and daclatasvir cleared HCV genotypes 1b and 3 in mice. Fluoxazolevir combined with glecaprevir and pibrentasvir was also effective in clearing multidrug-resistant HCV replication in mice. Fluoxazolevir may be promising as the next generation of combination drug cocktails for HCV treatment.
Main
Hepatitis C virus (HCV) is a positive-sense, single-stranded, 9.6-kb virus in the Flaviviridae family that infects over 70 million people worldwide1. HCV is one of the leading causes of liver cirrhosis, hepatocellular carcinoma and liver failure2. Once exposed to the virus, patients may remain asymptomatic for months, thereby impeding the treatment-seeking process3. Since interferon was first tested in the 1980s, the cure rate of HCV has steadily improved with the development of direct-acting antivirals (DAAs)4,5. A combination of first-generation DAAs with PEGylated interferon-α and ribavirin was first approved in 2011, elevating the cure rate to nearly 90% from about 50% with just PEGylated interferon and ribavirin4,5. Current DAA combination regimens are more effective with fewer side effects and have a higher barrier to drug resistance, improving the sustained virological response (SVR) rate to more than 90%4,5.
Despite this progress, there are still areas of unmet need in HCV therapy. Many individuals infected with HCV do not have access to existing treatments because of high costs6. Also, DAA therapy is less effective in difficult-to-treat patients, such as genotype 3 HCV infection with or without cirrhosis7. New and unusual subtypes (non-1a/1b, 3b, 4r) have also been discovered in patients from Asia and Africa and are less responsive to the current pan-genotypic regimen, sofosbuvir/velpatasvir, with a 50% SVR8,9,10. DAAs and other commonly used drugs have undesirable side effects and drug-drug interactions11. Many current treatment durations are lengthy at 12-24 weeks, although, in some cases, 8 weeks may suffice12. Shorter treatment durations may reduce costs and improve compliance. Finally, the emergence and transmission of HCV strains with multidrug resistance-associated substitutions (RASs) are a growing concern since they are less responsive to DAA retreatment13,14,15,16. In some studies, the response to retreatment is lower than 50% due to these multidrug RASs14. HCV reinfection occurs invariably in the transplant setting; effective preventive treatment, such as the use of hepatitis B immunoglobulin in preventing hepatitis B virus reinfection after liver transplant, would be valuable17. Therefore, new antivirals are needed to improve treatment efficacy and shorten treatment duration.
We previously identified a promising aryloxazole-based series of HCV entry inhibitors, which have a structural scaffold different from other described HCV entry inhibitors18. After further structure-activity relationship optimization, we identified the compound 18a (NCGC00351982 or fluoxazolevir) as the lead candidate for preclinical development based on the best combined profile of efficacy, cytotoxicity and in vitro absorption, distribution, metabolism and excretion (half maximal effective concentration (EC50) = 0.0188 μM, 50% cytotoxic concentration (CC50) = 13.0 μM, selectivity index CC50/EC50 > 600)19. We report here the mechanism of action of fluoxazolevir, including in vitro efficacy against various HCV genotypes, synergy with U.S. Food and Drug Administration-approved HCV drugs, in vivo pharmacokinetics in mice, rats and dogs, and efficacy in a humanized chimeric mouse model against HCV genotype 1b, 2a or 3 infection.
Results
Fluoxazolevir inhibits HCV fusion with hepatic cells
In a previous study, fluoxazolevir (Fig. 1a) was shown to target the entry step of the HCV life cycle using an HCV pseudoparticle assay19. To confirm fluoxazolevir's role in inhibiting HCV entry, a time-of-addition assay was performed20. Bafilomycin A1, a vacuolar-type H+-ATPase inhibitor, (S)-CCZ, a previously identified HCV late entry inhibitor20 and sofosbuvir, an NS5B polymerase inhibitor, were used as controls. Overall, fluoxazolevir showed a similar pattern of HCV inhibition to that of (S)-CCZ (Fig. 1b). Both fluoxazolevir and (S)-CCZ displayed potent inhibition similar to the continuous treatment when added either simultaneously or 2 h before infection. When fluoxazolevir and (S)-CCZ were administered 1 h after infection, both compounds were still effective in inhibiting infection. Bafilomycin A1 behaved similarly, but when added either 2 h before or 1 h after infection, it was much less effective, suggesting a more transient effect. In contrast, sofosbuvir was completely ineffective when the treatment was administered 2 h before infection but very potent when added simultaneously or any time after infection. The time-of-addition assay confirmed that fluoxazolevir targets the entry stage of the HCV life cycle.
A membrane fusion assay was performed to define whether fluoxazolevir targets viral fusion or another viral entry step (Fig. 1c)21. To prevent premature endosomal acidification, and consequently HCV entry, 10 mM of NH4Cl was added in all solutions throughout the assay22. Cell receptor binding was synchronized when high-titre HCV with NH4Cl was added to cells for 3 h at 4 °C (ref. 23). Forced HCV internalization and fusion with cytosolic lysosomes were then triggered by changing the overall pH of the medium to pH 5 for 5 min. After the pH shift, cells were incubated at 37 °C for 3 h, washed, cultured in regular media without NH4Cl for 72 h and then analysed for infection rate.
In the fusion assay (Fig. 1c,d), compounds were added at various times to test for specificity in inhibiting viral fusion. In protocols I and II, bafilomycin A1 behaved similarly to the dimethyl sulfoxide (DMSO) control treatment. As expected, the artificial lowering of the cytosolic pH overcame the block of endosomal acidification by bafilomycin A1, thus allowing HCV fusion to occur23. In contrast, HCV infection only increased minimally by 1.6-fold after the pH shift in the fluoxazolevir treatment group, which was lower than the increases of the DMSO (6.5-fold) and bafilomycin A1 (3.5-fold) groups. This finding indicates that fluoxazolevir blocks viral fusion within the endosomes even under an acidic environment. In protocol III, both fluoxazolevir and bafilomycin A1 failed to inhibit HCV infection since the compounds were added after the viral fusion step. Altogether, fluoxazolevir specifically inhibits the fusion step of HCV entry.
Fluoxazolevir binds to the HCV envelope protein 1 (E1)
To investigate the target of fluoxazolevir, a fluoxazolevir-diazirine-biotin (fluoxazolevir-DB) probe was synthesized (Extended Data Fig. 1a). Fluoxazolevir-DB showed inhibition against HCV infection in a dose-dependent manner with an EC50 of 1.19 µM (Extended Data Fig. 1b) and was stable at room temperature and under ambient light with slow decomposition after a few days (Extended Data Fig. 1c,d). When performing the fluoxazolevir-DB cross-linking experiment with recombinant HCV E1/E2 proteins under ultraviolet irradiation, the activated cross-linked product was identified to be the E1 protein by western blot with anti-E1 antibody (Fig. 1e). Under various control conditions, such as fluoxazolevir-DB without ultraviolet activation, DMSO and a sample with excess fluoxazolevir (200 µM) to compete against fluoxazolevir-DB (2 µM), the E1 protein was not detected. A similar ultraviolet cross-linking experiment was performed with fluoxazolevir-DB (5 µM) and Huh7.5.1 cells infected with high-titre chimeric genotype 1a HCV and showed specific cross-linking of fluoxazolevir-DB to E1 (Fig. 1f).
Fluoxazolevir RASs in E1
To further study the mechanism of action and genetic barrier to drug resistance of fluoxazolevir, an in vitro drug-induced resistance selection assay was performed24. Fluoxazolevir resistance emerged after 21 passages (Supplementary Fig. 1) compared to 11 passages for the NS5A inhibitor daclatasvir (Supplementary Fig. 2), indicating that fluoxazolevir may have a higher genetic barrier to resistance than daclatasvir. Amplified viruses from some of these passages (wells A1, B1, C1, E1, G1 and H1) showed a substantial shift of fluoxazolevir dose-response curves (an increase of EC50 > twofold), indicating the generation of the fluoxazolevir RASs (Extended Data Fig. 2). It is not clear why amplified viruses from other passages (wells D1 and F1) did not show any substantial resistance to fluoxazolevir. It is possible that certain RASs may have been less fit and promptly reverted to the wild-type (WT) sequence in the final amplification passage, when the compound was not added. Sequencing of the core, E1 and E2 regions of the viral isolates at the last stage of each selection passage identified various potential RASs (Fig. 2a,b), which were validated in the amplified viral stock. In the selection assay with daclatasvir (Supplementary Fig. 2), a common RAS (NS5A F28C) was found15,25, supporting the validity of this assay.
Mutations were then introduced individually into the HCV WT genome to confirm their resistance against fluoxazolevir. The mutant viral clones replicated similarly (no more than 20% difference) to the HCV WT clone (Extended Data Fig. 3a,b). Analysis of infectious virus production in the culture supernatant showed that most RAS-containing viruses produced similar levels of infectious virus in comparison to the HCV WT except for two E2 mutants: M405V and P616A, which produced somewhat lower infectious viral titres, and V414A, which produced more infectious virus (Extended Data Fig. 3a,c). The E1 RASs showed minor to moderate resistance (Fig. 2c,d and Extended Data Fig. 4). Among them, A274S, I374T, D382E and V414A exhibited notable resistance with the EC50 shifting from 36.7 nM against the HCV WT to 201, 242, 169 and 176 nM, respectively. Many of the mutations clustered in the E1 fusion peptide sequence, supporting the concept that fluoxazolevir targets the HCV fusion process. Two E1 mutations (I374T and D382E) occurred outside the fusion peptide and showed resistance (Fig. 2c). Mutations in the E2 protein (T395A, M405V, P616A) were also detected but they all occurred in the presence of validated resistant E1 mutations; when tested individually, they did not show much resistance (Fig. 2c and Extended Data Fig. 4).
Fluoxazolevir inhibits HCV chimeric infection
Dose-response assays of fluoxazolevir were performed against all chimeric HCV-Renilla luciferase (RLuc) genotypes including 1a, 1b, 2b, 3a, 4a, 5a, 6a and 7a (Fig. 3)26 and compared to the J6/JFH1 HCV-RLuc (genotype 2a). Fluoxazolevir was generally effective against all HCV genotypes and reached a maximum inhibition close to 100% at concentrations below notable toxicity. Fluoxazolevir showed genotypic variations in efficacy with varying EC50 values. It was most effective against HCV 2a and 2b, followed by 3a and 6a, all within sub-μM EC50 values. Fluoxazolevir also displayed little to no cytotoxicity, with CC50 > 20 μM in primary human hepatocytes, MT-4 cells, HepG2 cells and peripheral blood mononuclear cells (Extended Data Fig. 5), and approximately 12 µM in Huh7.5.1 cells (Fig. 3).
Fluoxazolevir synergizes with other anti-HCV drugs
To explore the potential combination of fluoxazolevir with currently available anti-HCV drugs, we tested the synergistic antiviral effects of fluoxazolevir with human interferon-α, ribavirin, daclatasvir, sofosbuvir and simeprevir (NS3/4A protease inhibitor). Two commonly used programs to calculate synergy, CalcuSyn and MacSynergy II, were applied27. CalcuSyn calculates combination indices (CIs) by analysing the inhibitory effects near the EC50 values for each drug28, while MacSynergy II uses the Bliss independence model29. Both programs use different definitions to determine the level of synergy; thus, each program provides a different but complementary profile of synergistic analysis. Drug combinations were added in a dose-dependent manner to determine whether the inhibitory effects of the treatment were synergistic, additive, equal or antagonistic to the inhibitory effects of each drug independently. CalcuSyn showed that fluoxazolevir was highly synergistic with all five selected antivirals while MacSynergy II demonstrated varying extents of synergism (Table 1).
Pharmacokinetic and toxicity studies in animal models
After single-dose administration in mice and rats, fluoxazolevir showed preferential localization in the liver with long t1/2 values for both intravenous and oral routes: 17-37 h in the plasma and 26-45 h in the liver (Extended Data Figs. 6a,b and 7). When fluoxazolevir was administered intravenously (3 mg kg−1), the volume of distribution at steady state (Vdss) was 137 l kg−1 and 12 ± 3 l kg−1 for CD-1 mice and Sprague Dawley rats, respectively. The high values of Vdss suggested that the compound penetrated tissues extensively. After oral administration (10 mg kg−1), the Cmax values in the plasma were 0.084 μM and 0.017 μM, the Cmax values in the liver were 34.4 and 39.9 μM and the liver to plasma area under the curve (AUC) ratios were 659 and 6,250 for CD-1 mice and Sprague Dawley rats, respectively (Extended Data Fig. 7). The oral bioavailabilities were 37 and 1.2% for CD-1 mice and Sprague Dawley rats, respectively, after a 10 mg kg−1 oral administration (Table 2).
In dogs, fluoxazolevir exhibited a similar pharmacokinetic profile as in rodents (Table 2 and Extended Data Fig. 6c), with a long t1/2 via intravenous (3 mg kg−1 dose; 29 h) and oral (10 mg kg−1 dose; 19 h) routes, high plasma clearance (67 ± 6 ml min kg−1), large Vdss (144 ± 15 l kg) and bioavailability of 14%. Analysis of urine samples collected for 10 d after the intravenous dosing showed a total renal excretion of 2.3 ± 0.4% of the administered dose.
A single dose of fluoxazolevir in these animals did not show any evidence of liver injury (alanine aminotransferase elevation) or other notable toxicity (Extended Data Fig. 6d). The maximum tolerable dose in CD-1 mice was determined by administering a single dose of 50, 100, 500 or 1,000 mg kg−1 fluoxazolevir by oral gavage with daily assessment of toxicity (body weight, observation, mortality and necropsy) for 3 d (Extended Data Fig. 8). No evidence of toxicity at any of those doses was observed.
Fluoxazolevir suppresses HCV infection in humanized chimeric mice
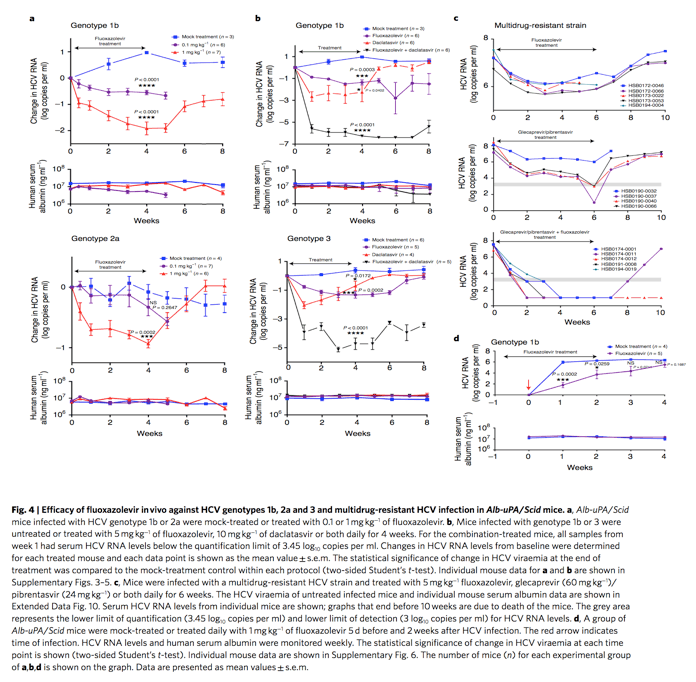
The antiviral effect of fluoxazolevir was tested in human hepatocyte-engrafted Alb-uPA/Scid chimeric mouse models infected with HCV genotypes 1b, 2a or 3. Fluoxazolevir was administered intraperitoneally daily for 4 weeks in two dosing groups for genotypes 1b and 2a (0.1 mg kg−1 and 1 mg kg−1) and one dosing group for genotype 3 (5 mg kg−1), and the animals were followed off-treatment for an additional 4 weeks. During treatment, viral RNA levels steadily declined for all genotype infections treated with fluoxazolevir (Fig. 4a and Supplementary Fig. 3) compared to the untreated mice. The 1 mg kg−1 dose was more effective in genotype 1b-infected mice, which decreased the viral RNA titre by about 2 log, than in genotype 2a-infected mice, which decreased the viral titre by about 1 log. The 5 mg kg−1 dosage for genotype 3-infected mice had a decrease in viral RNA titre of approximately 1.5 log (Fig. 4b and Supplementary Figs. 4 and 5). Throughout the course of treatment, there was no evidence of viral rebound, but RNA levels rebounded after treatment ended. No RASs were identified after sequencing the virus before and after treatment, suggesting a high barrier of drug resistance in vivo (Supplementary Table 1). Finally, there was no evidence of toxicity during the course of treatment (Extended Data Fig. 9).
Fluoxazolevir and daclatasvir combination therapy
Based on the synergy results and the demonstrated antiviral effects of fluoxazolevir in vivo, a 4-week combination therapy of fluoxazolevir and daclatasvir was conducted in humanized Alb-uPA/Scid mice infected with HCV genotype 1b or 3 to evaluate whether an SVR could be achieved. Monotherapy with daclatasvir was performed in comparison. The doses administered for fluoxazolevir and daclatasvir were 5 mg kg−1 intraperitoneally daily and 10 mg kg−1 orally daily, respectively. In the combination treatment of mice infected with both genotypes, the viral RNA levels in the serum rapidly decreased to undetectable levels without any evidence of emerging drug resistance and remained undetectable 4 weeks after stopping treatment, which is consistent with an SVR (Fig. 4b). On the other hand, daclatasvir monotherapy caused a rapid decline in viral levels, but the viraemia either never reached undetectable levels or rebounded, probably a result of emerging RASs. This study demonstrates that fluoxazolevir in combination with a DAA can achieve SVR against different HCV genotypes.
Human serum albumin levels were measured to monitor the engrafted human hepatocytes in all mice. During the entire course of treatment and follow-up, the human serum albumin levels remained relatively constant (Fig. 4a-d, Supplementary Figs. 3-5 and Extended Data Fig. 10), indicating that the reduction of HCV RNA was not caused by a loss of engrafted hepatocytes. One mouse from each treatment group infected by HCV genotype 1b showed a gradual decline of human serum albumin with time (Supplementary Fig. 4).
Fluoxazolevir is active against multidrug-resistant HCV
Mavyret, a combination of glecaprevir (a NS3/4A inhibitor) and pibrentasvir (an NS5A inhibitor), is a second-generation DAA regimen that is active against all HCV genotypes in vitro and in vivo and shows little or no loss of efficacy in commonly reported RASs30,31,32. Despite its high clinical efficacy, drug-resistant variants have been reported33. An HCV genotype 1b strain resistant to glecaprevir/pibrentasvir was generated in humanized chimeric mice infected with HCV genotype 1b by serial treatment with glecaprevir/pibrentasvir34. The virus contains NS3-D168E, a well-known NS3/4A RAS31, and multiple NS5A RASs (Q24R, R30E, P58S and A92K). Humanized chimeric mice infected with this virus respond poorly to glecaprevir/pibrentasvir treatment; interestingly, the RASs persist in the mice despite the absence of treatment34. NS5A-P58S has been reported in Mavyret-treated HCV patients with disease relapse33. NS5A-R30E has not been reported but other RASs affecting this residue are known32,35.
A 6-week combination therapy with fluoxazolevir and glecaprevir/pibrentasvir was conducted in humanized chimeric mice infected with this multidrug-resistant HCV strain. Fluoxazolevir was administered at a daily dose of 5 mg kg−1 while glecaprevir and pibrentasvir were administered at a daily dose of 60 mg kg−1 and 24 mg kg−1, respectively. In addition, fluoxazolevir or glecaprevir/pibrentasvir was administered separately as monotherapy groups. Another group of infected mice was untreated and monitored for viraemia, which showed steady levels during follow-up (Extended Data Fig. 10a). Fluoxazolevir-treated mice showed a 1-2 log gradual decline of HCV viraemia (Fig. 4c, upper panel), similar to what was observed in mice infected with HCV WT genotypes 1b, 2a or 3. In glecaprevir/pibrentasvir-treated mice, HCV viraemia declined by 2-5 log but never reached an undetectable level, except for 1 time point in mouse HSB0190-0037 (Fig. 4c, middle panel). All mice rebounded to pretreatment viraemia levels after fluoxazolevir or glecaprevir/pibrentasvir was stopped. In combination-treated mice, HCV RNA decreased rapidly to undetectable levels and remained below detectable levels throughout the duration of treatment, indicating the effectiveness of fluoxazolevir together with glecaprevir/pibrentasvir in suppressing this multidrug-resistant variant (Fig. 4c, lower panel and Extended Data Fig. 10b). After stopping treatment, all three surviving mice continued to show undetectable levels of HCV RNA for a week. With a longer follow-up of 4 weeks, one mouse died, one showed a viral rebound and the third continued to have an undetectable level of HCV RNA. Sequence analysis of the virus in the mouse with post-treatment relapse showed the same drug-resistant substitutions in NS3 and 5A as the inoculum virus (Supplementary Table 1).
Acute HCV infection is delayed by fluoxazolevir
Since fluoxazolevir targets viral entry, it may potentially serve as an HCV preventive treatment. To determine the effectiveness of fluoxazolevir as a preventive therapy for HCV infection, a 1 mg kg−1 daily treatment was administered intraperitoneally for 5 d before and 2 weeks after HCV genotype 1b inoculation (Fig. 4d and Supplementary Fig. 6). In the control group without treatment, viral RNA levels increased to about 6 log a week after infection while the viral RNA levels for the pretreated group gradually increased only to about 3 log. After stopping treatment, HCV RNA levels steadily increased for both groups; however, the viral RNA levels of the preventive group were still significantly lower than those of the control group. These data suggest that an entry inhibitor, such as fluoxazolevir, can partially prevent and hinder the progression of de novo HCV infection in vivo.
Discussion
In generating fluoxazolevir-resistant HCV clones, we identified multiple mutations in the putative E1 fusion loop, a sequence spanning between amino acids 264 and 290 (refs. 36,37), which confer drug resistance to fluoxazolevir. Two of the mutations were also induced by (S)-CCZ, another HCV fusion inhibitor (M267V and F291L)38, but the other RASs were unique to fluoxazolevir (A274S and F291V). Therefore, selection of fluoxazolevir-resistant substitutions in the fusion peptide of E1 shows that fluoxazolevir blocks HCV entry by interrupting the viral fusion process. Fluoxazolevir-DB also binds directly to E1 through ultraviolet-activated cross-linking, further supporting that it interacts with E1 to prevent viral entry. Two E1 RASs (I374T and D382E) outside the fusion peptide in the distal transmembrane domain, potentially play a role in anchoring the transmembrane domain to the plasma membrane and the fusion process39,40. Both mutants also showed lower viral fitness in comparison to the WT, probably due to a disruption of structural integrity in this hydrophobic domain41.
E2 interacts closely with E1 as a heterodimer40 and may play a role in the fusion process42 in addition to its interaction with host entry factors. Thus, E2 mutations may contribute to resistance against fluoxazolevir. F345V/V414A mutations in HCV genotype 3a (S52 strain) were reported to enhance the release of infectious virus particles and confer resistance against interferon-α43. Increased viral fitness could account for the apparent drug resistance against the V414A mutant. Analyses of the replicating and infectious capacities of the described mutants show that many of them are less infectious than the WT, suggesting that these RASs are less fit and may not persist once the drug is removed.
Fluoxazolevir can achieve complete inhibition against all seven HCV chimeric genotypes in vitro with varying efficacies. Fluoxazolevir is most active against HCV genotypes 2a and 2b, which is not unexpected since HCV genotype 2a was used to discover fluoxazolevir19. The in vivo studies where fluoxazolevir is effective against genotypes 1b, 2a and 3 without the emergence of RASs support the broad genotypic coverage of fluoxazolevir. The pharmacokinetic studies demonstrated a favourable profile with high liver concentrations, long t1/2 and reasonable oral bioavailability. With the dose administered, it was possible to achieve drug concentrations (>10 µM) in the liver that are substantially higher than the EC50 against all HCV genotypes without substantial toxicity.
Efficacy studies in humanized Alb-uPA/Scid mice showed that fluoxazolevir significantly suppressed HCV RNA levels during either monotherapy or combination therapy with daclatasvir. In mice infected with HCV genotypes 1b, 2a and 3, fluoxazolevir monotherapy decreased viral RNA significantly with no evidence of viral RNA rebound or generation of RASs. Daclatasvir monotherapy showed a greater decline in viral levels initially, which rebounded later during treatment, suggesting the emergence of RASs. In genotype 1b- or 3-infected mice treated with a combination of fluoxazolevir and daclatasvir, viral RNA levels were mostly below the detectable limit throughout the entire treatment period and 4 weeks after stopping treatment. Some of the post-treatment mice showed a detectable but not quantifiable HCV viraemia at some points after stopping treatment, which may represent residual non-infectious viral genomes after clearance since it does not lead to a substantial viral rebound.
A potent entry inhibitor, especially in combination with other DAAs, can minimize resistance and shorten treatment duration. Recent development of an entry inhibitor, myrcludex B, against chronic hepatitis D44, is a case in point for this therapeutic strategy. Entry inhibitors may also potentially prevent or delay HCV graft reinfection in liver transplantation. Recent use of HCV-positive organs in HCV-negative recipients has presented a unique opportunity for pre-emptive therapy with DAAs to prevent infection45,46. Potentially, fluoxazolevir could be used at a higher dose or in combination with another DAA for this purpose.
In summary, we have shown that fluoxazolevir inhibits HCV entry by blocking membrane fusion of viral endosomes, which is also the mechanism of action for other recently described entry inhibitors, such as chlorcyclizine, flunarizine and 4-aminoquinoline derivatives47,48,49. Our preclinical studies support fluoxazolevir as a promising candidate for the next generation of drug cocktails for HCV treatment. It is synergistic with U.S. Food and Drug Administration-approved HCV antivirals, active against all HCV genotypes in vitro, has preferential localization in the liver, can clear various HCV strains in a humanized mouse model and has potential to delay or prevent acute HCV infection. Since the viral fusion structure and process is relatively conserved50, it is also tempting to speculate that fluoxazolevir may have broader antiviral activities against other viruses.
| |
| |
| |
|
|
|