| |
Novel Antiretroviral Agents
|
| |
| |
Download the PDF here
Download the PDF here
Mary C. Cambou1 & Raphael J. Landovitz1,2
Current HIV/AIDS Reports Feb 2020
Conclusions
Of the novel ART agents discussed and showing promise in recent trials, only ibalizumab has current FDA approval. Preliminary data from the first phase Ib trial for the subcutaneous CAI GS-6207 suggested potential for extended-duration dosing [15]. While HIV developed resistance to previous MI, results from a phase IIb trial for boosted GSK2838232, a second-generation MI, demonstrated promising antiviral activity [23]. A phase I trial to evaluate a transdermal implant of islatravir, a highly potent, first-in-class NRTTI, maintained concentrations above the target PK threshold after 3 months, with implications in both PrEP and cART [28]. Extended-duration oral formulations of islatravir also have the potential to dramatically impact HIV treatment and prevention. Fostemsavir, the first-in-class attachment inhibitor, is available for compassionate use in HTE adults with MDR HIV-1. While China and Russia approved the fusion inhibitor ABT and the next-generation NNRTI elsulfavirine for use in cART, respectively, FDA approval in the USA has not been granted.
The treatment options with extended-duration dosing are particularly attractive, although tolerability and the PK tail, with the potential to select for resistant quasispecies absent continued dosing as levels wane, are of concern. Further clinical and post-marketing studies are needed to monitor the safety and efficacy of these new ART mechanisms. Agents with non-daily dosing requirements have the potential to be more successful in achieving and maintaining virologic suppression in PLWH challenged by adherence to daily oral tablets, including via the delivery in non-medical or directly observed therapy settings. The ongoing development of novel antiretroviral agents is needed to improve the HIV treatment cascade, with goals of improving the lives of PLWH, and reducing secondary transmission in an effort to end the HIV epidemic globally.
Abstract
Purpose of Review
Combination antiretroviral therapy (cART) has had dramatic effects on morbidity and mortality for persons living with HIV (PLWH). Despite significant progress in treatment efficacy, tolerability, and reducing pill burden, new agents are needed to address issues of resistance, drug-drug interactions, end organ disease, and adherence. This review covers novel ART agents recently approved or in development.
Recent Findings
Capsid inhibitors (CAI) demonstrate high potency and potential for extended-duration dosing in pre-clinical trials. While previous maturation inhibitors (MI) were hampered by issues of drug resistance, a recent phase IIa trial for a second-generation MI demonstrated promising antiviral activity. A phase I trial to evaluate a transdermal implant of islatravir, a nucleoside reverse transcriptase translocation inhibitor (NRTTI), maintained concentrations above the target pharmacokinetic threshold at 12 weeks. The attachment inhibitor fostemsavir is available in the USA for compassionate use in multi-drug-resistant (MDR) HIV.
Summary
New antiretroviral agents show promise for both extended-duration dosing and MDR HIV.
Introduction
Advances in modern antiretroviral therapy (ART) have dramatically reduced morbidity and mortality for PLWH. The past three decades have witnessed significant advances in potency, safety, and pill burden with the treatment of HIV since the introduction of zidovudine monotherapy in 1987 [1,2,3,4]. An estimated 60% of the 38 million PLWH globally receive ART. Owing to its high barrier to resistance and excellent tolerability, the integrase strand transfer inhibitor (INSTI) class, and dolutegravir in particular, has replaced the non-nucleoside reverse transcriptase inhibitor (NNRTI) class as the preferred third agent in cART: the updated 2018 World Health Organization recommendations list dolutegravir with a two nucleoside reverse transcriptase inhibitor (NRTI) backbone as the preferred first-line ART regimen [5].
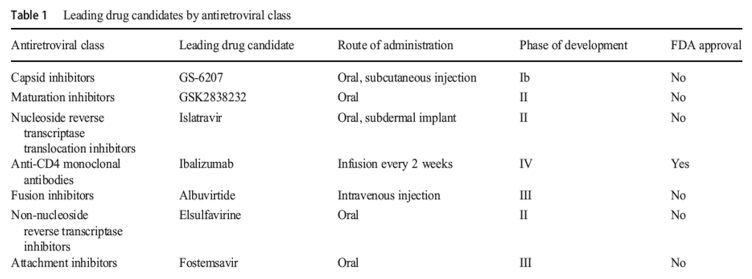
Despite these advances in ART leading to high rates of virologic suppression and tolerability, drug-induced and transmitted resistance and variable adherence to daily tablets remain challenges to the field. The development of novel ART agents and preparations with extended-release formulations and limited toxicity profiles are needed to address this gap in the HIV treatment toolbox. Here, we review novel agents in development or recently approved from the following ART classes: capsid inhibitors (CAI), maturation inhibitors (MI), nucleoside reverse transcriptase translocation inhibitors (NRTTI), anti-CD4 monoclonal antibodies (mAb), fusion inhibitors (FI), attachment inhibitors (AI), and non-nucleoside reverse transcriptase inhibitors (Table 1).
HIV Combinectin GSK3732394: A Long-Acting Inhibitor With Multiple Modes of Action
GSK3732394: a Multi-specific Inhibitor of HIV Entry
Importance: There continue to be significant unmet medical needs for patients with HIV-1 infection. One way to improve adherence and decrease the likelihood of drug-drug interactions in HIV-1-infected patients is through the development of long-acting biologic inhibitors. Building on a bi-specific inhibitor approach targeting CD4 and gp41, a tri-specific molecule was generated with three distinct antiviral activities. The linkage of these three biologic inhibitors creates synergy that offers a series of advantages to the molecule. The addition of human serum albumin to the tri-specific inhibitor could allow it to function as a long-acting self-administered treatment for patients with HIV infection. This molecule is currently in early clinical trials. Abstract: Long-acting antiretrovirals could provide a useful alternative to daily oral therapy for HIV-1-infected individuals. Building on a bi-specific molecule with adnectins targeting CD4 and gp41, a potential long-acting biologic, GSK3732394, was developed with three independent and synergistic modes of HIV entry inhibition that potentially could be self-administered as a long-acting subcutaneous injection. Starting with the bi-specific inhibitor, an α-helical peptide inhibitor was optimized as a linked molecule to the anti-gp41 adnectin, with each separate inhibitor exhibiting at least single-digit nanomolar (or lower) potency and a broad spectrum. Combination of the two adnectins and peptide activities into a single molecule was shown to have synergistic advantages in potency, the resistance barrier, and the ability to inhibit HIV-1 infections at low levels of CD4 receptor occupancy, showing that GSK3732394 can work in trans on a CD4+ T cell. Addition of a human serum albumin molecule prolongs the half-life in a human CD4 transgenic mouse, suggesting that it may have potential as a long-acting agent. GSK3732394 was shown to be highly effective in a humanized mouse model of infection. GSK3732394 is currently in clinical trials.
NIAID Launches First Clinical Trial to Test Antibody-Drug Combination for Long-Acting HIV Treatment - (01/27/20)
RC07-523LS broadly neutralizing antibody (bNAb), The first clinical trial to test the combination of a long-acting anti-HIV drug plus a powerful HIV antibody as a long-acting treatment for people living with HIV has begun.
A Study of Long-Acting Cabotegravir Plus VRC-HIVMAB075-00-AB (VRC07-523LS) to Maintain Viral Suppression in Adults Living with HIV-1 A Multicenter Trial of the AIDS Clinical Trials Group (ACTG) - A5357 - (01/24/20)
Pharm Wk: Early Safety, Tolerability and Pharmacokinetic Profile of GSK2838232, a Novel 2nd Generation HIV Maturation Inhibitor, as Assessed in Healthy Subjects - (06/20/17)

Capsid Inhibitors
A conical capsid core, comprised of approximately 250 capsid protein (CA) hexamers, surrounds the HIV RNA genome [6, 7]. After fusion of the HIV membrane with a CD4+ T cell, the capsid core transports the reverse transcription complex to the target nucleus and undergoes an uncoating process by which the CA hexamers disassemble [6, 8]. While the mechanism is poorly understood, reverse transcription of the RNA relies on uncoating [8]. The viral DNA contained in the capsid core, referred to as the pre-integration complex, shuttles through the cytoplasm via another poorly elucidated process involving CA. During virion maturation, CA is cleaved from the Gag polyprotein precursor to form the capsid core. The integral role of CA in both replication and virion maturation makes it a promising target for drug development.
While in vitro studies on capsid-targeting inhibitors began in 2003 [9], clinically relevant drugs did not reach presentation in the public domain until recently. Data for GS-CA1 was first presented at the conference on retroviruses and opportunistic infections (CROI) in 2017 as the first-in-class picomolar capsid inhibitor [10]. GS-CA1 and GS-6207 (also known as GS-CA2, an analog of GS-CA1) bind to the linker connecting the N-terminal and C-terminal domains that form the capsid protein. The dual mechanism of action is similar to PF74, another capsid inhibitor first introduced in 2010, although with higher affinity for the binding site [10]. GS-CA1 showed promise in pre-clinical trials owing to its high potency, including against resistant HIV-1 strains [9, 11]. However, five amino acid mutations at the GS-CA1 binding site were identified in in vitro resistance studies, raising concern about naturally occurring polymorphisms potentially compromising activity [10, 12]. In a cross-sectional, single-center study of 137 treatment-naïve PLWH, deep sequencing of the CA region was evaluated at baseline prior to treatment initiation. In 132 of the samples that were successfully sequenced, none of the amino acid substitutions identified in in vitro resistance studies was isolated [12], allaying concerns about naturally occurring resistance.
One of the main advantages of the CAI class, in addition to a high barrier to resistance, is the long-acting potential due to low predicted hepatic metabolic clearance based on cryopreserved hepatocyte models. The combination of prolonged half-life and aqueous solubility suggests the possibility of monthly subcutaneous dosing. Pharmacokinetic studies in rats and dogs demonstrated levels anticipated to be therapeutic at 12 weeks after administration of a single subcutaneous dose of GS-6207 [13]. Currently, subcutaneous GS-6207 is being investigated in the first phase Ib randomized controlled trial to evaluate antiviral activity in both treatment-naïve and treatment-experienced adults with HIV-1 [14]. Preliminary data demonstrated a mean 1.8 to 2.2 log10 decline in HIV-1 RNA at day 10 after a single subcutaneous dose, and there were no grade 3 or 4 adverse events requiring discontinuation [15].
Safety and antiviral activity over 10 days following a single dose of subcutaneous GS-6207, a first-in-class, long-acting HIV capsid inhibitor in people living with HIV
Maturation Inhibitors
The virion maturation process plays an important role in the infectivity of HIV-1. Normally, HIV-1 protease cleaves the junction between the CA and spacer peptide 1 (SP1) of the Gag polyprotein, resulting in a mature virion [16]. Bevirimat (also known as PA-457) was introduced in 2007 as the first maturation inhibitor (MI) to target the last step in the Gag cleavage process by binding to the CA-SP1 cleavage site, leading to the release of immature virions. Initially, bevirimat showed promise in phase I trials of treatment-naïve adults with HIV-1 [17]. However, a phase IIb study to assess the antiviral efficacy of bevirimat monotherapy in treatment-experienced adults with HIV-1 found that 55% of the participants had < 0.5 log10 viral load reduction after 15 days due to naturally occurring Gag polymorphisms near the CA-SP1 site [18]. Subsequent studies confirmed the presence of Gag polymorphisms conferring resistance [19], leading to the eventual discontinuation of bevirimat development.
Data for GSK3532795 (formerly known as BMS-955176), a second-generation MI, was first presented publicly at CROI in 2015 [16]. While structurally similar to bevirimat, GSK3532795 demonstrated high potency against several HIV-1 clades in vitro, including those with the Gag polymorphisms that compromised bevirimat potency [20, 21]. However, a phase IIb trial to evaluate GSK3532795 in combination with tenofovir and emtricitabine found treatment-emergent resistance in 15 of the 153 participants at virologic failure, including 10 cases of reverse transcriptase M184 mutations (two M184I, one M184I/V, and seven M184V) [22]. In addition to higher rates of treatment-emergent resistance compared with the efavirenz-based control, there was a 4-fold higher rate of gastrointestinal adverse effects [22]. Given these concerns, development of GSK3532795 was also subsequently suspended.
GSK2838232 is another second-generation MI that demonstrated potent activity across HIV-1 subtypes in vitro [23]. In a phase I dose-escalation trial to evaluate GSK2838232, the half-life was observed to increase 100% to 34 h when boosted with ritonavir, and achieved the target plasma exposure with higher doses of 200 mg daily compared with doses of 20, 50, and 100 mg [24]. The results of a proof of concept, dose-ranging phase IIa trial to evaluate the safety, pharmacokinetics (PK), and antiviral activity of GSK2838232 boosted with cobicistat were recently presented publicly at CROI [23]. In 33 treatment-naïve adults, a dose proportional response was observed with increasing doses of GSK2838232, with a mean 1.7 log10 decline in HIV-1 RNA from baseline after 10 days of 200 mg orally daily (compared with a mean 0.67 log10 decline in HIV-1 RNA for those treated with 20 mg orally daily). There were no polymorphisms associated with decreased GSK2838232 activity at baseline. Additionally, no serious clinical adverse events or grade 3 to 4 lab abnormalities were observed [23]. Additional safety and efficacy studies are not yet in the field.
Nucleoside Reverse Transcriptase Translocation Inhibitors
In contrast to NRTI, the nucleoside reverse transcriptase translocation inhibitor class has dual mechanisms of action: a 4′-ethynyl group inhibits translocation, and in combination with a 3′-hydroxyl group, results in chain termination [25]. Islatravir (formerly MK-8591) is the first-in-class NRTTI. In a recent in vitro potency study, the islatravir inhibitory quotients for both wild-type and NRTI-resistant HIV subtypes were found to be significantly higher than tenofovir disoproxil, tenofovir alafenamide, zidovudine, and lamivudine [26]. In a phase I trial of oral islatravir monotherapy in treatment-naïve adults with HIV-1, a single dose of 0.5 mg led to a mean 1.2 log10 decline in HIV-1 RNA at day 7 [25]. At steady state, concentrations of the active islatravir metabolite in both rectal and vaginal tissue were comparable with peripheral blood mononuclear cells, and the half-life ranged from 120 to 177 h [25]. Prolonged half-lives have been observed in animal studies as well. In PK studies of parenteral islatravir in rodents, continuous duration of drug release (owing to the recycling of drug accomplished by translocation inhibition) was observed after 180 days [27]. In macaque pre-exposure prophylaxis (PrEP) rectal challenge models, all macaques that received weekly islatravir were protected against simian immunodeficiency virus [28]. Its efficacy in animal models and long-acting potential suggest that it may be an option for extended-duration PrEP. A phase IIa randomized control trial (RCT) to evaluate once-monthly oral islatravir as PrEP in humans is currently underway [29].
A subdermal islatravir implant consisting of removable polymer was recently developed [30]. In PK studies of islatravir-eluting implants in rodents, there was an initial burst of drug release, followed by plasma steady state within days. The plasma levels were maintained above the expected trough concentration for once-weekly oral dosing at 12 months [31]. Similar results were observed in macaques treated with islatravir implants, suggesting feasibility in humans. A recent double-blind RCT in 16 healthy individuals evaluated the PK profile and tolerability of 54 mg and 62 mg implants [30]. At 12 weeks, both implants reached concentrations above the target PK threshold, and the 62 mg implant is projected to maintain concentrations above the PK threshold at 12 months. All adverse events were considered mild to moderate, and no clinically significant differences were observed between the placebo and treatments groups. However, there were higher reports of implant site erythema and pain in the 62 mg implant group compared with the 54 mg implant group. The tolerability and favorable PK profile of an islatravir-eluting implant have implications for both PrEP, and if combined with additional agents, cART [30].
Anti-CD4 Monoclonal Antibodies
Effective viral entry depends on the HIV envelope (Env) glycoprotein 120 (gp120) docking with the extracellular CD4 receptor [32, 33]. The CD4-gp120 complex subsequently undergoes conformational changes that facilitate viral fusion [32]. Non-competitive anti-CD4 monoclonal antibodies (mAb) that inhibit HIV entry were first described in the 1990s. While anti-CD4 mAb prevent viral fusion by binding to the surface opposite the major histocompatibility complex site, they are neither immunogenic nor obstruct normal immune activity [34].
Ibalizumab (formerly TNX-355) is a humanized mAb that binds to the N-terminal of domain 2 of the CD4 receptor [35]. When compared with broadly neutralizing antibodies PG9 and VRC01 in a large PK study, ibalizumab was more potent (by 10-fold) and neutralized the majority of 116 HIV strains at 50% inhibition of infection [35]. However, naturally occurring resistance to ibalizumab through loss of a V5 glycan loop in the envelope was documented, consistent with previous phase Ib trails that demonstrated treatment-emergent resistance via a similar mechanism [36]. In a phase IIb trial of 113 adults with MDR HIV that compared an infusion of 800 mg of ibalizumab every 2 weeks to 2000 mg every 4 weeks with an optimized background regimen, a > 1.0 log10 decline in HIV-1 RNA was observed for greater than 80% of participants in both treatment groups [37].
In 2018, the results of a phase III trial of ibalizumab infusion with an optimized background regimen in MDR HIV-1 [38] prompted US regulatory approval for treatment of MDR HIV. In an open-label trial of 40 adults with MDR HIV-1 and a viral load of > 1000 copies/mL, the mean viral load decrease was 1.1 log10. After 25 weeks, 43% of the participants had a viral load less than 50 copies/mL. Eight of the ten participants with virologic failure or rebound developed loss of the V5 loop function, as previously described [38].
Fusion Inhibitors
The HIV Env complex consists of three glycoprotein 41 (gp41) and gp120 subunits. As noted above, docking of gp120 to the CD4 receptor induces a conformational change whereby the gp41 ectodomain folds into a six-helix bundle (6HB) that facilitates viral and CD4 membrane fusion [39, 40]. Enfuvirtide (T20), a 36-amino acid lipopeptide derived from the C-terminal heptad repeat region of gp41, inhibits viral fusion by binding to the N-terminal heptad repeat region, thus preventing complete 6HB formation [39, 41]. Enfuvirtide is the only currently FDA-approved fusion inhibitor in the USA. However, its twice daily subcutaneous dosing owing to its low in vivo half-life (max 4.35 h), poor tolerability, need for refrigerated storage, and low barrier to resistance makes it an infrequently used component of cART [39].
Albuvirtide (ABT) is a novel 3-maleimimidopropionic acid-modified peptide derived from C34, another C-peptide with a similar mechanism of action to T20 [39]. ABT binds strongly to albumin, resulting in an extended half-life of 12 days [42]. In a phase II trial to evaluate weekly injections of ABT with ritonavir-boosted lopinavir (LPV/r), there was a mean 1.9 log10 and 2.2 log10 decline in HIV-1 RNA from baseline at week 7 in the 160 mg and 320 mg ABT groups, respectively [42]. Interim results from TALENT, a phase III non-inferiority trial comparing weekly ABT with LPV/r to two NRTIs with LPV/r in treatment-experienced PLWH, were presented in 2016 [43]. At week 48, only 66.0% of the triple-drug arm had HIV-1 RNA < 50 copies/mL compared with 80.4% of the ABT arm. In 5 patients with HIV-1 RNA > 400 copies/mL at 24 and 48 weeks in the ABT arm, no gp41 mutations were identified and there was no statistically significant difference observed in adverse events between the two groups [43]. In 2018, China approved the use of ABT for the treatment of HIV-1 [44]. While the final results of the TALENT trial have not been published, a phase II trial to evaluate ABT in combination with 3BNC117 (a broadly neutralizing antibody) as a long-acting maintenance combination in virologically suppressed PLWH is currently underway in the USA [45].
Attachment Inhibitors
Fostemsavir (BMS-663068, the pro-drug of temsavir) is a first-in-class attachment inhibitor that prevents HIV entry into the CD4 T cell by binding to the viral envelope gp120. The 96-week results of the BRIGHTE study, a multinational phase III, two-cohort (non-randomized and randomized) clinical trial to evaluate the efficacy and safety of fostemsavir in heavily treatment-experienced (HTE) adults with MDR HIV-1, were publicly presented at IAS in 2019 [46]. HTE adults failing their current regimen with HIV-1 RNA > 400 copies/mL, and unable to construct a viable regimen, with at least one fully active approved ART agent, were included in the randomized cohort. Of 203 participants who received fostemsavir 600 mg twice a day plus their current regimen, there was a median 1.0 log10 decrease in HIV-1 RNA at day 8 in participants with a baseline HIV-1 RNA > 1000 copies/mL [46]. In the intention-to-treat analysis, 60% of the randomized cohort reached HIV-1 RNA levels < 40 copies/mL, and the mean increase in CD4 count was 205 cells/mm3 at week 96. However, 21% in the randomized cohort reported grades 2–4 drug-related adverse events (AE), of which 3% were serious drug-related AE. Individuals with MDR HIV-1 with viral loads > 1000 copies/mL and unable to construct a viable regimen with currently approved ARV options are eligible for fostemsavir compassionate use [47].
Non-Nucleoside Reverse Transcriptase Inhibitors
The NNRTI class has evolved significantly since the Food and Drug Administration (FDA) approval of nevirapine in 1996. Elsulfavirine (VM-1500, the pro-drug of VM-1500A) is a next-generation, highly selective NNRTI [48]. In a phase Ib/IIa trial to evaluate the pharmacokinetics of VM-1500 in PLWH, the half-life of the VM-1500A metabolite was 7.4 and 5.4 days after 1 week of daily 20 mg and 40 mg doses of elsulfavirine, respectively [49]. A phase Ib trial to evaluate the safety and pharmacokinetics of oral, once-weekly elsulfavirine is currently underway [50].
The results of a 48-week, phase IIb multicenter RCT comparing daily elsulfavirine to efavirenz, combined with tenofovir disoproxil fumarate and emtricitabine, were presented in 2017 [51]. Among 120 treatment-naïve PLWH, 81% of the elsulfavirine arm had HIV-1 RNA < 50 copies/mL compared with 73.7% of the efavirenz arm at week 48. There were no cases of virologic failure in either arm. Elsulfavirine was well-tolerated: adverse events were reported in 36.7% of participants in the elsulfavirine arm compared with 77.6% of the efavirenz arm (p value < 0.0001). Five of the 60 participants enrolled in the elsulfavirine arm discontinued the study drug compared with 13 participants receiving efavirenz due to drug-related adverse events [51]. In 2017, Russia approved the use of elsulfavirine for the treatment of HIV, although it has not yet obtained regulatory approval in the USA.
Conclusions
Of the novel ART agents discussed and showing promise in recent trials, only ibalizumab has current FDA approval. Preliminary data from the first phase Ib trial for the subcutaneous CAI GS-6207 suggested potential for extended-duration dosing [15]. While HIV developed resistance to previous MI, results from a phase IIb trial for boosted GSK2838232, a second-generation MI, demonstrated promising antiviral activity [23]. A phase I trial to evaluate a transdermal implant of islatravir, a highly potent, first-in-class NRTTI, maintained concentrations above the target PK threshold after 3 months, with implications in both PrEP and cART [28]. Extended-duration oral formulations of islatravir also have the potential to dramatically impact HIV treatment and prevention. Fostemsavir, the first-in-class attachment inhibitor, is available for compassionate use in HTE adults with MDR HIV-1. While China and Russia approved the fusion inhibitor ABT and the next-generation NNRTI elsulfavirine for use in cART, respectively, FDA approval in the USA has not been granted.
The treatment options with extended-duration dosing are particularly attractive, although tolerability and the PK tail, with the potential to select for resistant quasispecies absent continued dosing as levels wane, are of concern. Further clinical and post-marketing studies are needed to monitor the safety and efficacy of these new ART mechanisms. Agents with non-daily dosing requirements have the potential to be more successful in achieving and maintaining virologic suppression in PLWH challenged by adherence to daily oral tablets, including via the delivery in non-medical or directly observed therapy settings. The ongoing development of novel antiretroviral agents is needed to improve the HIV treatment cascade, with goals of improving the lives of PLWH, and reducing secondary transmission in an effort to end the HIV epidemic globally.
|
|
| |
| |
|
|
|