| |
Targeting Inflammation to Reduce Atherosclerotic
Cardiovascular Risk in People With HIV Infection
|
| |
| |
Download the PDF here
HIV infection alters markers of vascular endothelial dysfunction through mechanisms which remain incompletely understood. As previously described, endothelial dysfunction is an important precursor of atherogenesis.
In vitro studies provide some evidence for the direct toxicity of viral proteins gp120 and Tat (transactivator of viral replication) to cardiac and vascular cell lines, suggesting a potential direct role of HIV replication in mediating endothelial dysfunction.48, 49 This likely does not fully explain the endothelial dysfunction observed in PLWH, given that markers of endothelial dysfunction remain higher in PLWH on cART with effective viral suppression.50 Finally, cART itself has been implicated in endothelial dysfunction and further confounds the mechanistic drivers of this process in PLWH
In addition to effects on the endothelium, lipid metabolism appears to be directly altered by HIV infection.53, 54 Before the rollout of cART, clinical studies of untreated HIV-positive individuals demonstrated lipid disorders, including increased triglycerides, increased LDL, and low high-density lipoprotein serum levels, in patients with advanced disease and severe immunosuppression.55, 56, 57 Chronic inflammation and immune activation may play a role in dyslipidemia observed in individuals with untreated HIV infection. In untreated PLWH, high levels of IFN (interferon)-alpha may decrease clearance of triglycerides, leading to elevated serum levels,55, 56 whereas low levels of serum high-density lipoprotein have been associated with elevated IL-6 and D-dimer levels.57 Treatment with cART generally leads to some improvement in low high-density lipoprotein cholesterol, but does not normalize the lipid profile in treated PLWH.58, 59 Several antiretroviral drug classes have also been implicated in altering lipid metabolism and increasing oxidative stress in PLWH, potentially independently contributing to increased cardiovascular risk (reviewed in Cunha60).
Anti-PCSK9 Monoclonal Antibodies: The Future Lipid-Lowering Agent of Choice?....http://www.natap.org/2016/AGE/AGE_49.htm
PCSK9 price-cut matchup is on, as Regeneron and Sanofi slash Praluent list tag 60%....http://www.natap.org/2019/newsUpdates/021519_02.htm
Heavy Popper Use in Gay MACS Cohort Linked to New Heart Disease, Cancer ......http://www.natap.org/2014/CROI/croi_73.htm
siRNA drug safely halved LDL cholesterol in phase 3 ORION-11....http://www.natap.org/2019/HIV/090919_03.htm
Efficacy and Safety of Bempedoic Acid in Patients WithHypercholesterolemia and Statin Intolerance
The main finding of the CLEAR Serenity trial is that bempedoic acid significantly reduces both LDL and hsCRP compared with placebo and is well tolerated in patients with statin intolerance. .....http://www.natap.org/2020/HIV/022420_01.htm
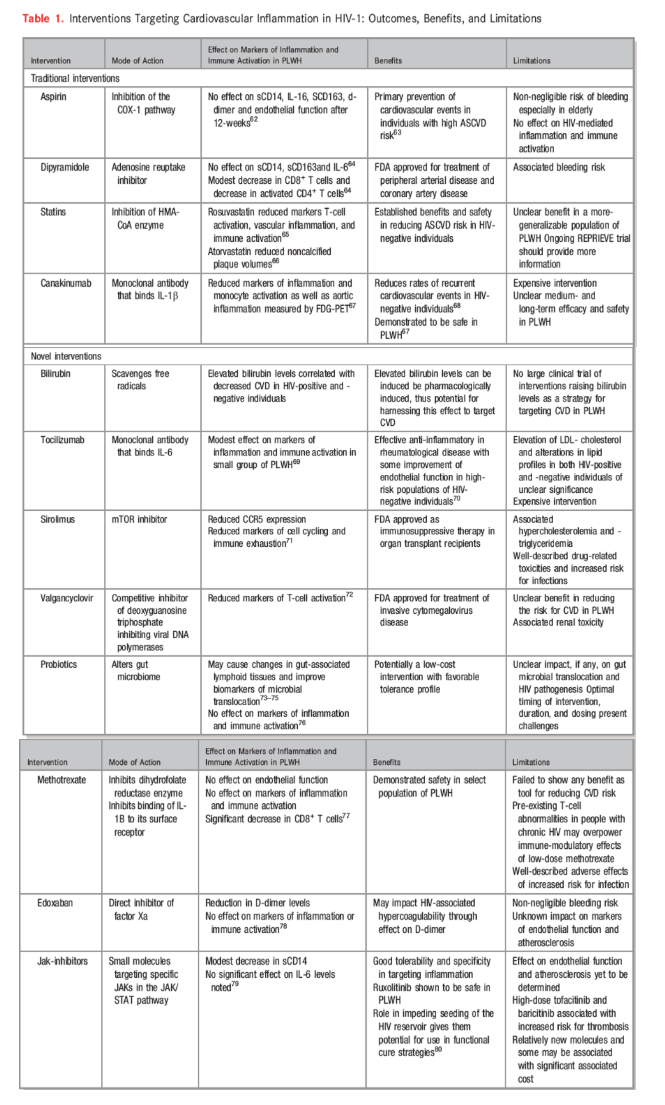
Table 1. Interventions Targeting Cardiovascular Inflammation in HIV-1: Outcomes, Benefits, and Limitations
Targeting Inflammation to Reduce Atherosclerotic Cardiovascular Risk in People With HIV Infection
Originally published 24 Jan 2020
Boghuma Titanji, MD, PhD; Christina Gavegnano, PhD; Priscilla Hsue, MD; Raymond Schinazi, PhD; Vincent C. Marconi, MD
Introduction
In almost 4 decades since the start of the HIV epidemic, the world has witnessed an unprecedented evolution of this disease from a debilitating and rapidly fatal syndrome to a chronic condition, effectively managed with combination antiretroviral therapy (cART). Today people living with HIV (PLWH) who receive treatment have nearly the same life expectancy as HIV-negative individuals.1 With the exception of 2 people cured by human stem-cell transplantation,2, 3 a widely applicable cure for HIV remains elusive and infection still requires lifelong therapy for PLWH. Although effective cART suppresses viral replication, inflammation and immune activation persist for PLWH and are driven by a combination of HIV-dependent and HIV-independent factors.4 These immune factors contribute to an excess of non-AIDS comorbidities in PLWH, including cardiovascular disease (CVD), frailty, malignancy, neurocognitive disease, osteoporosis, and renal and liver diseases.4 It is increasingly recognized that as the population of PLWH ages, targeting non-AIDS comorbidities is essential to effectively care for and treat this population.
CVD is the leading cause of death worldwide, accounting for 56.9 million deaths in 2016.5 The relative risk of CVD in PLWH is significantly higher than in HIV-negative controls, including: higher rates of acute myocardial infarction6 and increased risk for ischemic stroke,7 heart failure,8 and sudden cardiac death.9 In fact, it is estimated that the HIV-associated risk for CVD may be similar to that of traditional risk factors such as smoking, hyperlipidemia, diabetes mellitus, and hypertension.10 Despite several studies showing the higher risk of cardiovascular events in PLWH, the greatest challenge has been defining the overarching mechanisms by which HIV-mediated immune activation and chronic inflammation increase the risk for CVD.11 This has made it difficult to identify effective interventions to target and reduce cardiovascular risk in this population despite considerable efforts.
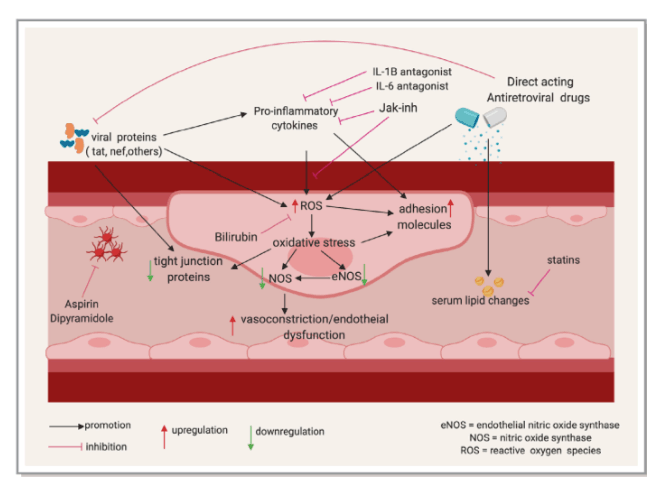
In this review, we examine the effects of HIV-associated inflammation and immune activation on the cardiovascular system with a focus on atherosclerotic CVD and discuss existing and proposed therapeutic strategies targeting inflammation to reduce CVD risk. The factors contributing to immune activation and CVD in PLWH are summarized in Figure 1 below.
Figure 1. Factors contributing to immune activation and cardiovascular disease in PLWH. Solid line arrows indicate a contributory effect; dotted line arrows represent a potential yet uncertain relationship; dotted terminal line indicates an inhibitory effect. ART indicates antiretroviral therapy; CMV, cytomegalovirus; HCV, hepatitis C virus; PLWH, people living with HIV. This figure was created using www.biorender.com software.
Mechanisms of Chronic Inflammation and Immune Activation in HIV Infection
Infection with HIV triggers a generalized activation of the immune system. This immune activation is both specific and nonspecific, involving several mechanisms.
Persistent Viral Production and Replication
During HIV infection, uncontrolled viral replication leads to progressive CD4+ T-cell decline, but also systemic inflammation and immune activation. In the SMART (Strategies for Management of Antiretroviral Therapy) trial, continuous suppression of HIV replication was associated with decreased risk of CVD when compared with intermittent therapy, suggesting a direct role for uncontrolled viral replication as a risk factor for CVD.12, 13 Subsequent studies have gone on to show an association between uncontrolled HIV replication and vascular endothelial dysfunction,14, 15 further highlighting the importance of cART to reduce cardiovascular risk in PLWH. This is especially relevant in Sub-Saharan Africa, which harbors 26 million PLWH with an estimated 40% of these individuals not on cART.16 The Ndlovu cohort study, founded in 2017, aims to provide insight into the burden of CVD and contribution of HIV infection in a rural area of Sub-Saharan Africa with high HIV prevalence.17 This study will include a total of 1000 HIV-positive and 1000 HIV-negative participants, with a male-to-female ratio of 1:1, and should provide useful information on the burden of CVD in this context as well as the implications of virological suppression with cART on the risk of CVD.17 In a prospective study of 82 treatment-naïve patients enrolled to initiate cART in the United States, treatment of HIV leading to virological suppression and immune reconstitution resulted in rapid improvement in brachial artery flow-mediated dilation (FMD), a measure of endothelial dysfunction.14 A smaller study by Baker et al also demonstrated that untreated HIV infection was associated with impaired arterial elasticity, measured by pulse waveform analysis, in both small and large vessels.
In elite controllers who maintain high CD4+ T-cell counts and suppress HIV viremia in the absence of cART, there is also evidence of innate immune activation and elevated serum markers for inflammation associated with increased risk for clinical events, higher rates of hospitalization, and CVD.18, 19, 20, 21, 22, 23 In a longitudinal study of 40 elite controllers, 98% of these individuals had measurable HIV RNA during a 16-month median follow-up period, often at levels higher than observed in patients receiving cART.24 These findings suggest that low-level viral replication, which may not be consistently detectable in elite controllers, may still be a significant driver of immune activation and inflammation in this group. These persistent T-cell activation levels in elite controllers may contribute to progressive CD4+ T-cell decline even in the absence of consistently detectable viremia,25 thus highlighting a potential role for treating elite controllers with cART in order to prevent non-AIDS-related comorbidities.19, 20, 26
T-Cell Activation, Dysfunction, and Depletion
CD4+ T-cell activation and depletion as well as CD8+ T-cell activation are central to the pathogenesis of HIV infection. Activated CD4+ T cells (CD38+ HLA-DR+) are significantly elevated in HIV-positive individuals compared with negative controls25 and decrease with early initiation of cART.27 Several studies have examined the association between CD4+ T-cell depletion and CVD and shown an independent association between lower CD4+ T-cell counts and increased cardiovascular risk.7, 28, 29, 30 In 1 study of 114 HIV-positive women compared with HIV-negative controls, women with HIV infection were found to have higher levels of CD38+ HLA-DR+ CD4+ T cells. This higher level of activated T cells was associated with increased carotid artery stiffness in this group.31 In the HIV-negative population, activated CD4+ T cells are frequently found in atherosclerotic lesions. It is plausible to speculate that in HIV infection, activated CD4+ T cells generate cytokines which are part of the cascade leading to atherosclerosis. A causal link is yet to be established between T-cell activation and risk for CVD.20, 32
Cytotoxic CD8+ T cells are important components of the adaptive immune response to HIV infection. Elevated levels of activated CD8+ T cells (CD38+ HLA-DR+) have been associated with subclinical carotid artery disease in HIV-positive individuals.31, 33 In the SATURN-HIV (Stopping Atherosclerosis and Treating Unhealthy bone with Rosuvastatin in HIV) trial, higher levels of CD8+ CD38+ HLA-DR+ T cells were associated with increased carotid media intima thickness (CIMT).33 The reasons behind persistent CD8+ T-cell activation, even among people on cART, are likely a combination of a persistent antiviral response to HIV infection as well as stimulation from other viral infections like cytomegalovirus. In a study of 93 HIV positive individuals compared with 37 HIV negative controls, cytomegalovirus-specific T-cell responses was shown to be independently associated with CIMT in HIV-positive individuals compared with negative controls.34
Monocyte/Macrophage Activation
There is considerable evidence to support that activation of monocytes and macrophages also enhances atherosclerosis in chronic HIV infection.35, 36, 37, 38 Activated monocytes and macrophages release soluble CD163 (sCD163) during inflammation, a serum marker associated with aortic inflammation and plaque volume in PLWH.35, 36, 39 Soluble CD14 (sCD14), another serum marker of macrophage activation, has also been shown to correlate with subclinical atherosclerosis in HIV-positive individuals,37 further implicating the innate immune response in the pathogenesis of CVD in chronic HIV infection. The central role of monocyte/macrophage activation in the pathogenesis of atherosclerosis in the general population is well recognized.40 Circulating monocytes adhere to vascular endothelium and transform into tissue macrophages, where they phagocytize oxidized low-density lipoprotein (LDL) molecules to become lipid-laden foam cells.41 Consequently, this triggers release of proinflammatory cytokines and adhesion molecules central to plaque formation and growth.41 The chronic state of inflammation and immune activation associated with HIV infection likely amplifies monocyte/macrophage inflammation and tissue damage in arterial plaque formation.
Effects of HIV-Mediated Immune Activation and Chronic Inflammation on the Cardiovascular System
Suppressing viral replication with cART significantly reduces immune activation for PLWH, but it does not eliminate it. PLWH have persistently higher levels of systemic inflammation when compared with HIV-negative controls both before and while receiving cART. This is reflected by higher levels of serum biomarkers (including hs-CRP [high-sensitivity C-reactive protein], IL [interleukin]-6, sCD163, sCD14, and D-dimer) observed in PLWH on cART compared with negative controls, driven by mechanisms previously discussed.35, 36, 38, 42, 43, 44 The resulting effects on the cardiovascular system are far reaching and likely explain the increased risk for cardiovascular events noted in this population, even when accounting for traditional cardiovascular risk factors.
Lipid Metabolism and Atherosclerosis
Vascular endothelial dysfunction and inflammation are central to the pathogenesis of atherosclerosis. A detailed discussion of atherosclerosis and lipid metabolism are beyond the scope of the current review. Briefly, in response to proatherogenic stimuli, such as oxidized LDL, the vascular endothelium produces nitric oxide, which alters vascular tone, upregulates production of chemokines and adhesion molecules, and increases endothelial permeability. This leads to increased deposition of low-density lipids in the vascular wall. Macrophage uptake of oxidized LDL induces transformation to foam cells, which are characterized by the further release of chemokines, proinflammatory cytokines, and growth factors.41 This causes progressive intimal thickening and plaque formation characteristic of atherosclerotic disease. If the plaque ruptures or erodes, this exposes the highly thrombogenic collagenous matrix and lipid core to the circulation. This, in turn, triggers platelet aggregation, fibrin deposition, and ultimately thrombus formation, which, at its worse, may ultimately cause obstruction of the vessel.41
HIV infection alters markers of vascular endothelial dysfunction through mechanisms which remain incompletely understood. As previously described, endothelial dysfunction is an important precursor of atherogenesis. In nonhuman primate studies, focal endothelial proliferation and increased subendothelial inflammatory cells were identified in thoracic aortas of simian immunodeficiency virus–infected macaques compared with negative controls.45 FMD of the brachial artery has been used as a measure of endothelial dysfunction and a predictor of CVD in the general population.46 To date, 1 prospective randomized controlled trial (RCT) has demonstrated a reduction in flow-mediated dilatation FMD in PLWH treated with cART.14 Furthermore, soluble VCAM-1 (vascular cell adhesion molecule-1), a marker for vascular endothelial activation, has also been shown to be elevated in HIV-positive patients, and levels were reduced by cART.47 In vitro studies provide some evidence for the direct toxicity of viral proteins gp120 and Tat (transactivator of viral replication) to cardiac and vascular cell lines, suggesting a potential direct role of HIV replication in mediating endothelial dysfunction.48, 49
This likely does not fully explain the endothelial dysfunction observed in PLWH, given that markers of endothelial dysfunction remain higher in PLWH on cART with effective viral suppression.50 Finally, cART itself has been implicated in endothelial dysfunction and further confounds the mechanistic drivers of this process in PLWH.51, 52
In addition to effects on the endothelium, lipid metabolism appears to be directly altered by HIV infection.53, 54 Before the rollout of cART, clinical studies of untreated HIV-positive individuals demonstrated lipid disorders, including increased triglycerides, increased LDL, and low high-density lipoprotein serum levels, in patients with advanced disease and severe immunosuppression.55, 56, 57 Chronic inflammation and immune activation may play a role in dyslipidemia observed in individuals with untreated HIV infection. In untreated PLWH, high levels of IFN (interferon)-alpha may decrease clearance of triglycerides, leading to elevated serum levels,55, 56 whereas low levels of serum high-density lipoprotein have been associated with elevated IL-6 and D-dimer levels.57 Treatment with cART generally leads to some improvement in low high-density lipoprotein cholesterol, but does not normalize the lipid profile in treated PLWH.58, 59 Several antiretroviral drug classes have also been implicated in altering lipid metabolism and increasing oxidative stress in PLWH, potentially independently contributing to increased cardiovascular risk (reviewed in Cunha60).
Hypercoagulability
Several markers of chronic inflammation have been linked with increased cardiovascular risk in the general population and in HIV-positive individuals.15, 61 IL-6 and hs-CRP are associated with increased mortality from CVD in PLWH and hs-CRP to increased CIMT in this population.8, 43 D-dimer, a product of the coagulation cascade and inflammation marker, is also increased in HIV infection and has been shown to have similar associations with CVD risk as IL-6 and hs-CRP.32 Besides being a marker of inflammation, high levels of D-dimer are associated with a procoagulable state. It is reasonable to hypothesize that a procoagulable state would increase the risk of cardiovascular events such as myocardial infarction (MI), stroke, and acute thrombosis.54, 61 A causal relationship between D-dimer elevation and cardiovascular events in PLWH has not yet been demonstrated and may simply be a reflection of the underlying chronic inflammatory state. The coagulation cascade remains, however, an attractive target for interventions aimed at reducing the risk of CVD in HIV infection.
Targeting Inflammation and Immune Activation to Reduce the Risk of CVD in HIV
Given the overwhelming evidence supporting the increased risk of CVD in patients in PLWH, several traditional and novel interventions are being investigated to reduce cardiovascular risk in this patient population (summarized in Table 1 below).
Directly Acting Modulators of Inflammation and Immune Activation
Antiplatelet Agents
Platelets play a key role in thrombus formation and atherogenesis and may also contribute to inflammation. Increased platelet activation is associated with an increased risk of cardiovascular events. As a result, antiplatelet therapy is recommended in those at highest risk for CVD and with low risk of bleeding. Aspirin, an antiplatelet drug that irreversibly inhibits the COX-1 (cyclooxygenase-1) pathways, has been the choice drug for this intervention based on clinical trial data.63
The effect of low-dose aspirin on markers of inflammation and immune activation for PLWH has been assessed in several observational studies and clinical trials. An open-label, uncontrolled study showed that 1 week of low-dose aspirin therapy in HIV-positive patients on cART reduced platelet aggregation and markers of T-cell/monocyte activation compared with untreated controls.81 This promising observation led to a prospective, blinded RCT with a longer intervention period of 12 weeks, comparing the effects of low-dose (100 mg) aspirin with a higher (300 mg) dose and a placebo on the markers of immune activation and inflammation in PLWH on cART. The larger trial did not show any effect of 100 or 300 mg of aspirin compared with placebo on levels of sCD14, IL-16, sCD163, D-dimer, or other markers of monocyte and T-cell activation. Endothelial function measured by FMD was also unaffected.62
Anti-inflammatory effects of aspirin are mediated by its ability to selectively block the COX-1 pathway, causing a downstream inhibition of prostaglandin and paracrine hormone production and propagating a broad variety of effects. These effects are highly nonspecific, which might explain the inability of aspirin to have a measurable effect on inflammation and immune activation for HIV-positive individuals, suggesting that more-targeted anti-inflammatory interventions may be required in order to observe an effect. Recent evidence has demonstrated the significant risk of major bleeding associated with the use of aspirin for primary prevention of CVD82 and highlighted the need to better define high-risk individuals with clinical atherosclerosis (such as those with a high coronary artery calcium score) in whom the benefits of this intervention may outweigh the bleeding risk.83 This is a consideration which is highly relevant in HIV-positive individuals, given that individuals in this group are more likely to have a pre-existing risk of bleeding features such as thrombocytopenia.
IL-1β Antagonists
IL-1β is an important inflammatory cytokine that drives the IL-6 signaling pathway. IL-1β plays a significant role in development of atherothrombotic plaque. It promotes coagulation, increases adhesion of leukocytes and monocytes to the vascular endothelium, and upregulates growth of smooth muscle cells.84, 85, 86 The CANTOS (Canakinumab Anti-Inflammatory Thrombosis Outcomes Study) trial was designed to test the inflammatory hypothesis of atherothrombosis by using canakinumab, a therapeutic monoclonal antibody which specifically targets IL-1β, to treat HIV-negative individuals.68 In patients with a previous MI and baseline elevated hs-CRP levels, 150 mg of canakinumab led to significantly reduced rates of recurrent cardiovascular events than placebo, independent of lipid lowering.68
The landmark findings from the CANTOS trial encouraged investigation of IL-1β antagonists to target inflammation and immune activation in PLWH. More recently, a nested case-control study and 183 HIV-negative controls from Denmark explored the IL-1 inflammation pathway as a predictor of first-time MI in PLWH. This study found that PLWH had higher levels of the IL-1 receptor antagonist at all time points leading to first-time MI, and these higher levels of IL-1 receptor antagonist were associated with a 1.5-fold increase in MI risk.87 These findings further support targeting the IL-1 activation pathway to reduce CVD for PLWH.
A small, open-label pilot study including 10 participants evaluated the safety and efficacy of canakinumab in targeting inflammation and immune activation for PLWH virologically suppressed with cART.67 This study showed that canakinumab therapy was safe and led to decreased levels of markers of inflammation and monocyte activation and also a reduction in aortic inflammation as measured by fluorodeoxyglucose positron emission tomography.67 Whereas these findings are encouraging, they are limited by the small numbers recruited and the short follow-up time. A larger and longer RCT (ClinicalTrials.gov Identifier: NCT02272946) is underway, which should help clarify the short- and medium-term efficacy and safety of canakinumab in PLWH. It is worth mentioning that the IL-1β antagonist, canakinumab, carries a treatment cost of approximately $200 000 per year for its current US Food and Drug Administration (FDA)-approved indications and recently failed to gain approval from the regulatory body for use in CVD prevention in HIV-negative individuals.
Low-Dose Methotrexate
Methotrexate is a chemotherapeutic and anti-inflammatory drug used to treat cancer and chronic rheumatic conditions such as rheumatoid arthritis (RA). The mechanism involves a competitive inhibition of the dihydrofolate reductase enzyme and also inhibition of IL-1β binding to its cell-surface receptor. Several large cohort studies have suggested the benefit of low-dose methotrexate therapy (LD MTX) for reducing atherosclerotic CVD risk.88, 89, 90, 91, 92 This prompted the investigation of LD MTX as a strategy to lower cardiovascular inflammation in a large RCT excluding PLWH. The CIRT (Cardiovascular Inflammation Reduction Trial), NCT01594333, aimed to randomize 7000 patients with previous MI and either type 2 diabetes mellitus or metabolic syndrome to LD MTX or placebo for 3 to 5 years. The trial was halted in early 2018 because of failure to show an effect on markers of inflammation or cardiovascular events compared with placebo.93 Hsue et al, in a smaller observational study, looked at the safety and impact of LD MTX on endothelial function in PLWH on cART.77 Although the intervention was safe, it did not have any demonstrable effects on endothelial function measured by brachial artery FMD or inflammatory biomarkers, but was associated with a significant reduction in CD8+ T cells.77
The failure of this intervention to show an effect again suggests that the pathways of inflammation in HIV infection likely differ significantly from those of inflammatory rheumatologic conditions, such as RA and psoriasis, for which methotrexate is effective treatment. Furthermore, the pre-existing T-cell abnormalities, even in virologically suppressed PLWH on cART, as included in this study, may overpower the immunomodulatory effects of LD MTX.77
IL-6 Antagonists
Elevated levels of IL-6 have been linked to an increased risk of cardiovascular events70 and predict morbidity and mortality for individuals living with HIV infection.94 The IL-6 antagonist, tocilizumab, is approved for treatment of RA.95 Given the central role of IL-6 as a driver of cardiovascular inflammation and atherogenesis, there has been interest in the use of IL-6 antagonists to reduce cardiovascular inflammation. In safety trials of tocilizumab for treatment of RA, lipid levels increased for some patients receiving the study drug,70, 96, 97, 98 raising a concern as to whether it would be beneficial or harmful as a potential agent to reduce the risk of CVD.
In 1 community-based, prospective study, tocilizumab was shown to improve endothelial function in a high-risk population despite its effect of increasing levels of LDL cholesterol.99 At the 2019 CROI (Conference on Retroviruses and Opportunistic Infections), a small RCT looking at the effects of tocilizumab among treated HIV-positive patients showed that the drug significantly altered lipid profiles for individuals on cART, but also seemed to modestly lower some markers of inflammation and immune activation in this patient population.69 Further studies are warranted to fully examine the impact of tocilizumab-induced lipid changes on CVD risk for PLWH.
Janus Kinase Inhibitors
The JAK (Janus kinase)/STAT (signal transduction and activator of transcription) pathway has been implicated as an important driver in the pathogenesis of inflammatory, hematological, and autoimmune conditions.100, 101, 102 Binding of a variety of ligands, such as cytokines, interleukins, and interferons, to cell-surface receptors causes activation of JAKs.
Activation of JAKs triggers cross-phosphorylation of these proteins and further downstream phosphorylation of a family of STAT proteins.100 Activation of the JAK/STAT pathway ultimately culminates in inducing transcription of genes for a range of proinflammatory and immunomodulatory cytokines.
Signaling pathways for important inflammatory cytokines TNF (tumor necrosis factor)-alpha, IL-6, and IL-1 have been successfully targeted by biological agents in treatment of autoimmune diseases and cancer. The success of tocilizumab (a monoclonal antibody targeting IL-6 for the treatment of RA) validated the potential for JAK1, JAK2, and TYK2 (tyrosine kinase 2) as viable therapeutic targets of inflammation.102, 103 Biological agents are highly successful and have revolutionized the treatment of autoimmune conditions and cancer in recent years, but also present significant limitations. Biologics are large proteins with the potential to be immunogenic, require administration by intravenous infusion or subcutaneous dosing, and treatments are associated with significant financial cost. Their use has also been associated with opportunistic infections, making them less attractive for use in PLWH.
Currently, 3 JAK inhibitors are FDA approved for clinical use-tofacitinib (RA and ulcerative colitis), baricitinib (RA), and ruxolinitib (myeloproliferative neoplasms)104, 105, 106-and at least 12 more drugs are being tested in clinical trials. Tolerability and specificity of JAK inhibitors present a unique advantage over traditional anti-inflammatory agents like NSAIDs. They are small molecules with oral bioavailability, an added advantage over biological agents. The earlier-generation JAK inhibitors, such as tofacitinib, which was the first of its class to be FDA approved, have the disadvantage of not being highly selective in its ability to block JAKs. Tofacitinib specifically blocks JAK3, but has a lower affinity for JAK1/JAK2. JAK3 blockade is associated with lower neutrophil and natural killer cell counts, which results in an increased risk for infections associated with its use.102 This observation led to a more-targeted approach in developing newer JAK inhibitors more specific for blocking JAK1/2. Ruxolitinib and its structural analog, baricitinib, specifically inhibit JAK1/2.107 Baricitinib has been demonstrated to be safe in pediatric populations in treatment of autoinflammatory interferonopathies108 and is under investigation for other indications in this population. Another important advantage is its low dose (2 or 4 mg) and minimal metabolism (poor substrate for cytochrome P450) and hence reduced potential for interaction with other drugs.109
When it comes to JAK inhibitors and their potential role in targeting HIV-related immune activation/inflammation and the implied CVD risk, these agents present an attractive option worthy of further exploration. Inflammatory cytokines, which are generated through the JAK/STAT pathway (IL-1, IL-6, IL-10, and IFN-alpha and -gamma), remain elevated in treated and untreated HIV-positive individuals and are associated with increased cardiovascular risk, morbidity, and mortality in this group. In addition, immunomodulatory cytokines, such as IL-2, IL-7, and IL-15, play an important role in triggering the JAK/STAT pathway and in recent reports have been implicated in proliferation of the HIV reservoir.110, 111 This presents a unique dual target for JAK inhibitors, both as potential modulators of chronic inflammation/activation and part of a functional cure strategy, by targeting expansion of the viral reservoir. A recent study by Gavegnano et al demonstrated the role of the JAK/STAT pathway in HIV persistence and showed the ability of ruxolitinib and tofacitinib to impede seeding and maintenance of the HIV reservoir,80 further supporting this theory.
Safety of JAK-inhibitors in HIV-positive people is currently being examined in clinical trials. The ACTG (AIDS Clinical Trials Group) 5336 open-label RCT recently assessed the safety, tolerability, and immunological activity of 5 weeks of ruxolitinib 10 mg in HIV-positive people on cART.79 This study showed that in a highly selected cohort of HIV-positive individuals, ruxolitinib was safe and led to a modest decrease in sCD14, though a nonsignificant reduction was noted in IL-6 levels for individuals receiving ruxolitinib compared with the control group.79 The findings of this trial provide support for future studies aimed at exploring the use of JAK-inhibitors to target residual inflammation and immune dysregulation in treated HIV infection. Cardiovascular outcomes have been examined in patients receiving tofacitinib for RA and psoriasis, with some indication that among treated individuals, it may lead to lower incidence of major cardiovascular events.105, 112 Of note, postmarketing reports of increased risk of thrombosis associated with high doses of tofacitinib and baracitinib to a lesser degree have been reported by the FDA and will need to be considered with any new clinical indications for this drug class.113, 114 The benefit of JAK inhibitors in specifically targeting cardiovascular inflammation should be examined in the near future.
Indirectly Acting Modulators of Inflammation and Immune Activation
Statins
These potent lipid-lowering drugs act by inhibiting the β-hydroxy β-methylglutaryl coenzyme A reductase enzyme and reduce the risk of CVD. Anti-inflammatory properties have also been attributed to statins, and landmark clinical trials have demonstrated their effects in reducing cardiovascular risk when used in both primary and secondary prevention for HIV-negative populations.115, 116 Among patients with HIV infection, statins have also been shown to reduce several markers of immune activation and CVD. Large retrospective studies suggest dramatic benefits of statins in reduction of all-cause mortality for PLWH.117 In contrast, within a large retrospective study of 3601 participants from the ALLRT (AIDS Clinical Trials Group Longitudinal Linked Randomized Trials) cohort, statins did not reduce time to non-AIDS-defining events.118
Statins exhibit antiviral properties against HIV in vitro119 but prospective and retrospective clinical studies on the effects of statins on HIV-1 RNA levels have yielded mixed results.120, 121, 122 An early small double blind placebo RCT that included 24 PLWH, high dose atorvastatin was found to decrease markers of T-cell activation, but had no effect on HIV-1 RNA levels.123 In the INTREPID (HIV-infected patients and treatment with pitavastatin vs pravastatin for dyslipidemia) study, an RCT comparing pitavastatin to pravastatin for treatment of dyslipidemia in PLWH, pitavastatin was associated with a reduction in markers of monocyte activation (sCD14) and arterial inflammation (oxidozed LDL and lipoprotein-associated phospholipase 2).124 More recently, a medium-size study with 303 PLWH on long-term suppressive cART examined the relationship between markers of immune activation/inflammation and viral persistence. With the exception of increasing IP-10 (IFN-γ-inducible protein 10), statin therapy in this cohort did not significantly lower markers of inflammation (IL-6, neopterin, sCD14, sCD163, and TNF-alpha) and did not have an effect on the viral reservoir.125
In the SATURN-HIV RCT comparing 10 mg of rosuvastatin to placebo for 48 weeks, rosuvastatin showed a reduction in markers of monocyte and T-cell activation for HIV-positive participants receiving antiretroviral therapy.65 CIMT progression was also found to be slower in the intervention group compared with placebo, and this effect was greatest in study participants who had the highest levels of inflammatory biomarkers.126, 127 A second RCT by Lo et al showed dramatic reductions in noncalcified plaque volumes measured by computed tomography angiography for patients with treated HIV infection receiving 20 or 40 mg of atorvastatin.66 Whereas some studies suggest that more-potent statins will be beneficial in reducing the risk of CVD in PLWH otherwise at low risk,127 other studies have failed to demonstrate an effect on immune activation/inflammation,125 raising questions as to the true effect of statins for PLWH.
The ongoing multicenter REPRIEVE trial will assess whether pitavastatin compared with placebo will reduce risk of CVD in a more-generalizable population of PLWH receiving cART, more representative of a "real world scenario."128
Participants are being enrolled based on the American College of Cardiology/American Heart Association atherosclerotic CVD risk score and LDL cholesterol level, with the goal of identifying individuals most likely to benefit from this intervention.128, 129 Whether the cardiovascular risk reduction attributed to statins for PLWH is attributable to a reduction in LDL cholesterol level versus a reduction in inflammation and immune activation or a combination of both remains unclear, and subanalyses from this trial are also planned with the aim of addressing this question.129 The REPRIEVE trial, which plans to recruit >7000 participants in 120 sites across 11 countries, should hopefully provide some mechanistic insights into how statins mediate CVD risk reduction among PLWH.
Valganciclovir
HIV infection is also indirectly responsible for stimulating the immune system through reactivation of latent forms of other viruses, including cytomegalovirus and Epstein–Barr virus.130 Depletion of CD4+ T cells during HIV infection may lead to a decreased ability of the immune system to control these persistent herpes viruses, allowing their reactivation and replication.131 Reactivation of cytomegalovirus during HIV infection has been shown to be associated with higher levels of cytomegalovirus-specific T-cell responses in coinfected individuals.54 In a WIHS (Women's Interagency HIV Study) cohort study of women living with HIV, higher serum titers of cytomegalovirus immunoglobulin G were associated with having a subclinical lesion in the carotid arteries.130 In the HIV-negative population, cytomegalovirus has been isolated from atherosclerotic lesions132 and shown to predict mortality in patients with coronary artery disease.133 Cytomegalovirus is a relatively ubiquitous pathogen with the ability to infect endothelial cells and smooth muscle cells.134 As a result, it has been cited as a possible pathogen for atherosclerosis,135 although there is a paucity of evidence to support a direct causal relationship. A small prospective study explored the role of targeting cytomegalovirus replication in PLWH with valganciclovir. In the valganciclovir-treated arm, reduced T-cell activation was noted in HIV-positive people on cART72; however, it remains unclear whether this translates to an overall decrease in CVD risk over time.
Probiotics
Dietary supplementation with probiotics as a strategy to alter the gut microbiome and reduce CVD for HIV-negative individuals has been extensively investigated in several studies (reviewed in Ettinger et al136). A meta-analysis of 13 RCTs including 485 participants showed that a probiotic-rich diet reduces LDL and total cholesterol levels in individuals with high, borderline, and normal cholesterol levels.137 Gut microbial translocation during HIV infection contributes to immune activation and chronic inflammation.37, 138, 139 Targeting this microbial translocation to reduce inflammation for PLWH is an attractive proposition, especially considering the potential collateral effects of reducing non-AIDS chronic conditions associated with HIV infection. There is some evidence, both in vivo and ex vivo, that probiotics may improve local and systemic inflammation in patients with inflammatory bowel disease.140 In chronically simian immunodeficiency virus–infected macaques, the combination of cART plus a probiotic led to an increase in gut mucosal CD4+ T cells and antigen-presenting cell frequency and function as well as reduced fibrosis.141
Several studies of probiotics for PLWH have also reported changes in gut-associated lymphoid tissues and improvement in biomarkers of microbial translocation.73, 74, 75 More recently, an RCT assessed the effect of probiotics on inflammation and immune activation in treated HIV infection.76 In this study, 47 participants were randomized to receive the probiotic, Visbiome ES, for 24 weeks compared with 46 participants in the placebo arm. Addition of probiotics to cART did not alter markers of inflammation and immune activation, had no effect on lipid profiles, and showed a modest effect on gut microbiome populations.76
Use of probiotics as a potential intervention to target inflammation and immune activation for PLWH poses unique challenges. The lack of an effect in the recent RCT may be attributable to the wrong target, the wrong population, or inadequate dosing or choice of probiotics. HIV infection appears to have its most significant impact on the gut in the early stages of infection, and as a result we may be less likely to observe a notable effect for interventions targeting chronically infected patients who have been on cART and are virologically suppressed. Nonetheless, this intervention, if demonstrated to be beneficial, would present a preventative option that is affordable, easy to administer, and with few adverse effects.
There is a need for more studies to clarify a potential role for probiotics in PLWH. It is unclear to what degree the pathogenesis of HIV infection in the gut can truly be altered by the use of these agents. Improving the microbial composition of the gut may not necessarily reverse its leakiness and the immune dysfunction at that interface. Furthermore, the changes in microbial composition of the gut may not be permanent once the probiotic is withdrawn.
Bilirubin
Bilirubin has been shown to have antiatherogenic characteristics attributed, in part, to its antioxidant properties.142 Individuals with Gilbert syndrome, who have a hereditary inability to conjugate bilirubin and elevated levels of unconjugated bilirubin as a result, have lower rates of stroke, coronary artery disease, and other inflammatory conditions.143 This property has been studied as a possible intervention to reduce the risk of CVD for HIV-negative individuals with diabetes mellitus. Through the use of pharmacological interventions, which increase serum bilirubin levels, several studies have shown a decrease in CVD with elevated bilirubin levels in patients with diabetes mellitus.144, 145, 146
More recently, analyses of 96,381 participants in the Veterans Aging Cohort Study, one-third of whom were HIV positive, showed that an elevated bilirubin level is inversely correlated with CVD among both HIV-positive and -negative individuals.147 Bilirubin elevations are common during treatment with the protease inhibitor, atazanavir. In a prospective RCT on CIMT, elevated bilirubin in study participants receiving atazanavir appeared to favorably influence CIMT progression rates.148 These findings raise the interesting prospect of harnessing the protective effect of elevated bilirubin to reduce cardiovascular risk in PLWH.
Mammalian Target of Rapamycin Inhibitors
These drugs inhibit the mTOR (mammalian target of rapamycin), which is an important cellular kinase mediating growth, cellular metabolism, and proliferation.149 The mTOR inhibitor, sirolimus, is frequently used for immunosuppression to reduce organ rejection in solid organ transplant patients.150 A retrospective analysis of the use of sirolimus in HIV-positive renal transplant recipients suggested that this drug may lower levels of proviral HIV DNA in CD4+ T cells.151 Recently, the potential role of sirolimus in modulating immune function and the HIV reservoir was investigated in a small, prospective, open-label single-arm study including 32 participants. Sirolimus was associated with a reduction in CCR5 (C-C chemokine receptor type 5) expression and T-cell markers of cell cycling and immune exhaustion; however, there was a high discontinuation rate attributed to associated drug toxicities and adverse effects.71 mTOR inhibitors are associated with considerable adverse effects, which may outweigh any potential benefits they have in targeting immune activation to reduce CVD risk or the viral reservoir in PLWH. A study investigating CVD risk in 1812 liver transplant recipients who received sirolimus as part of initial immunosuppression (sirolimus cohort) found that sirolimus was associated with hypertriglyceridemia and –cholesterolemia, but did not increase the risk of MI or other CVD compared with a nonsirolimus control group.152 Considering that the sirolimus cohort has more baseline cardiovascular risk factors and nearly double the 10-year cardiovascular risk of the control group in this study, the investigators argue that a similar incidence of CVD events in both the sirolimus and nonsirolimus group is suggestive of a cardioprotective effect of sirolimus in liver transplant recipients.152 More studies are needed to further clarify the role of sirolimus in CVD and as adjunctive therapy for modulating immune dysregulation in PLWH.
Factor Xa Antagonists
As discussed earlier, elevated D-dimer levels have been linked to a higher incidence of non-AIDS-related morbidity and death for PLWH.32, 153, 154 It is hypothesized that HIV-associated coagulopathy may contribute to disease by amplifying inflammatory pathways in addition to direct effects from thrombotic occlusions. Coagulation activity may drive immune activation by protease-activated receptors with downstream effects on the function of vascular endothelial surfaces.155, 156, 157, 158 Targeting the coagulation cascade as a way of reducing inflammation and immune activation for PLWH is an attractive area for investigation, especially considering potential implications for cardiovascular risk reduction and other non-AIDS-related morbidities.
Factor Xa is a key activator of protease-activated receptors 1 and 2, inhibition of which may reduce inflammation along vascular surfaces. The anti-inflammatory properties of factor Xa inhibitors pertaining to vascular inflammation in the absence of HIV infection have been reported in several studies.159, 160, 161, 162 In the TACTICAL HIV (Targeted Anticoagulation Therapy to Reduce Inflammation and Cellular Activation in Long-term HIV Disease) study, a crossover RCT, safety and efficacy of 30 mg of edoxaban (an antagonist of factor Xa) were assessed in participants with treated HIV infection.78 Whereas edoxaban led to significant reduction in D-dimer levels, it had no effect on markers of inflammation and immune activation.78 Use of edoxaban was also associated with a higher bleeding risk. In preliminary results presented at CROI 2019, the investigators did not specifically assess markers of endothelial function and atherosclerosis. These results provide promising leads, but also highlight the need for more studies to better understand the mechanisms of HIV-associated coagulopathy. The failure to notice an effect on inflammation and immune activation suggest that some of these mechanisms of coagulation may be distinct from the drivers of inflammation in PLWH. Other coagulation factors, notably factor IIa (thrombin), have been implicated in vascular inflammation, and targeting this factor with its direct inhibitor dabigatran improved endothelial function and reduced atherosclerosis in mice.163 This strategy has not been explored in the setting of HIV infection and presents an interesting avenue for future strategies targeting the coagulation cascade.
Adenosine Reuptake Inhibitors
Targeting adenosine reuptake has recently been explored as a way of targeting HIV-associated chronic inflammation and cardiovascular risk. Adenosine has potent immunoregulatory effects in response to cellular stress triggered by tissue damage and inflammation.164 Increased production of adenosine in the extracellular environment leads to downstream signaling effects initiated by the binding of adenosine to its cellular receptors.165 This culminates in increased production of cAMP within the cell and a dampening of immune response.166 Dipyramidole inhibits cellular reuptake of adenosine and raises its extracellular levels. It is also approved by the FDA as an antiplatelet agent for treatment of peripheral arterial disease and coronary artery disease.167 Macatangay et al, in a recent RCT, hypothesized that increasing extracellular levels of adenosine through use of dipyramidole could potentially dampen chronic inflammation and reduce cardiovascular risk in HIV-positive individuals.64
To test this hypothesis, they conducted a placebo-controlled, 2-arm, crossover pilot study in which dipyramidole was administered to 17 HIV-positive individuals on suppressive cART compared with 18 HIV-positive individuals who received placebo. Markers of endothelial dysfunction and inflammation were assessed after 12 weeks of treatment.64 Following the treatment period, there were no significant changes in sCD14, sCD163, or IL-6 levels in the intervention and control arms of the study. They did, however, note a modest decrease in activated CD8+ T cells as well as a significant decrease in activated CD4+ T cells in pooled analyses. There was no significant difference in FMD between treatment and control groups at the end of the study period.64
The small decrease in activated T cells following treatment with dipyramidole suggests that alterations in adenosine metabolism may contribute to T-cell activation in HIV-positive individuals who are treated and virologically suppressed. However, the lack of an effect on markers of inflammation and endothelial dysfunction indicates that adenosine metabolism may not play a significant role in monocyte/macrophage activation in PLWH and thus only partly explains the degree of residual inflammation and immune activation observed in chronic HIV infection. Similar to the other interventions discussed in this review, the selection of a healthy group of HIV-positive individuals with immune reconstitution and virological suppression may limit the ability to detect a measurable effect of the intervention.
Future Directions and Conclusions
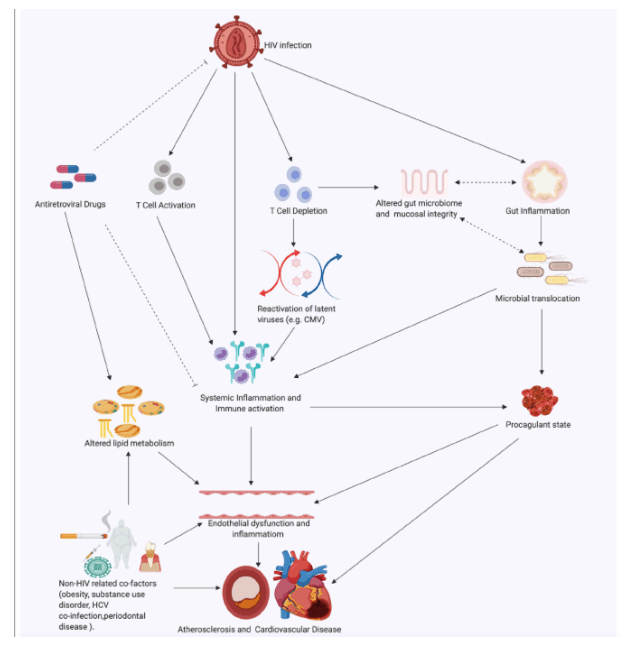
The role of HIV infection as an independent predictor for CVD is now well established, but the mechanisms by which this occurs remain to be fully defined. There is a growing body of evidence suggesting that chronic inflammation and immune activation, the direct effects of HIV proteins, and treatment with cART may be important in the pathogenesis of CVD among PLWH compared with their HIV-negative counterparts. The degree to which each of these factors contribute to the pathogenesis and pathways of CVD remain uncertain, hence the challenge in identifying clear therapeutic targets. One plausible theory is that the aforementioned factors create an environment of increased oxidative stress at the endovascular interface, leading to increased endothelial dysfunction and damage, which triggers atherogenesis and its consequent nefarious effects. This theory supports exploring strategies which aim to directly reduce inflammation and immune activation and oxidative stress by targeting reactive oxygen species. Whether these strategies would be sufficient in reversing the risk of CVD in PLWH or need to be combined with traditional interventions targeting other factors remain to be seen (see Figure 2 and Table 2168, 169, 170, 171, 172, 173, 174, 175 below).
Figure 2. Pathogenesis of endothelial damage attributable to HIV infection and its treatment: Viral proteins, inflammatory cytokines, and cART all lead to increase in reactive oxygen species (ROS). Oxidative stress induces endothelial nitic oxide synthase (eNOS) uncoupling leading to decreased nitric oxide (NO) availability. These events trigger endothelial dysfunction which is an important early step in endovascular damage and atherogenesis. This figure was created using www.biorender.com software. cART indicates combination antiretroviral therapy; IL-1β, interleukin-1 beta; IL-16, interleukin-16; Jak-inh, Janus kinase inhibitor.
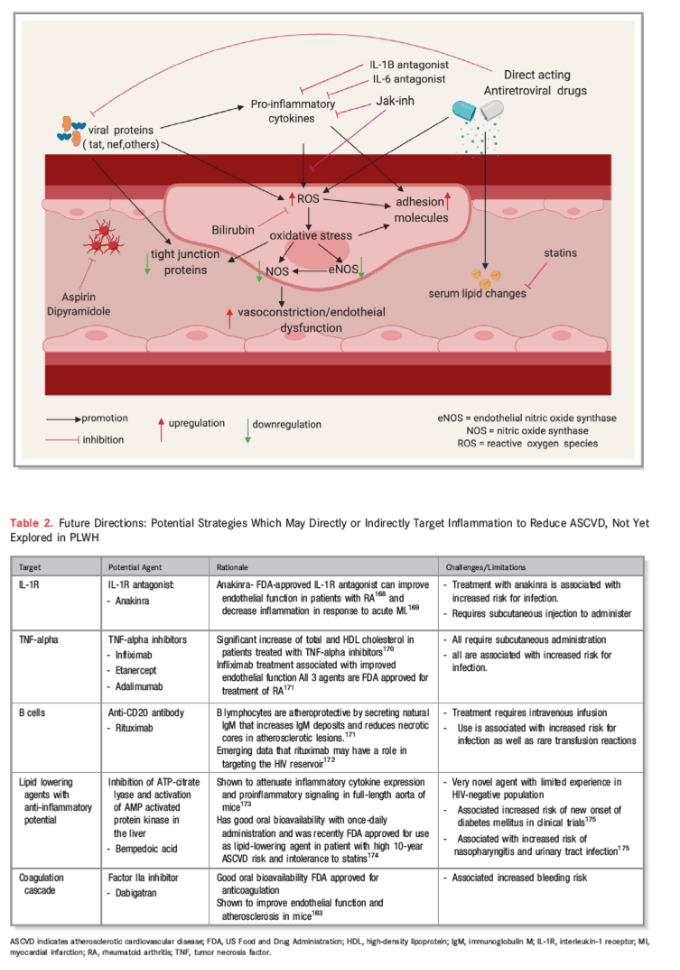
So far, robust RCT data that support targeting inflammation to reduce CVD risk in HIV-positive individuals are limited, with most of the observational data discussed in this review yielding mixed results. These studies have almost exclusively included relatively healthy virologically suppressed HIV-positive individuals on cART and may explain the limited ability to detect clear benefits of the interventions tested. Furthermore, larger studies encompassing the full spectrum of PLWH are needed to ascertain that therapies inhibiting inflammatory response in individuals with HIV will bring positive effects rather than deleterious ones. The currently available evidence highlights the need for studies aimed at better understanding the mechanisms of CVD in HIV, defining clear intervention targets, identifying clinically relevant markers to measure response, and the subset of patients most likely to benefit from these interventions.
|
|
| |
| |
|
|
|