| |
Long-Acting Cabotegravir and Rilpivirine for
Maintenance of HIV-1 Suppression ATLAS
|
| |
| |
Download the PDF here
Download the PDF here
NEJM March 19 2020 - S. Swindells, J.-F. Andrade‑Villanueva, G.J. Richmond, G. Rizzardini, A. Baumgarten, M. Masiá, G. Latiff, V. Pokrovsky, F. Bredeek, G. Smith, P. Cahn, Y.-S. Kim, S.L. Ford, C.L. Talarico, P. Patel, V. Chounta, H. Crauwels, W. Parys, S. Vanveggel, J. Mrus, J. Huang, C.M. Harrington, K.J. Hudson, D.A. Margolis, K.Y. Smith, P.E. Williams, and W.R. Spreen
Every-2-Month Maintenance CAB + RPV Noninferior to Monthly Dosing for 48 Weeks - (03/09/20)
Abstract
Background
Simplified regimens for the treatment of human immunodeficiency virus type 1 (HIV-1) infection may increase patient satisfaction and facilitate adherence.
Methods
In this phase 3, open-label, multicenter, noninferiority trial involving patients who had had plasma HIV-1 RNA levels of less than 50 copies per milliliter for at least 6 months while taking standard oral antiretroviral therapy, we randomly assigned participants (1:1) to either continue their oral therapy or switch to monthly intramuscular injections of long-acting cabotegravir, an HIV-1 integrase strand-transfer inhibitor, and long-acting rilpivirine, a nonnucleoside reverse-transcriptase inhibitor. The primary end point was the percentage of participants with an HIV-1 RNA level of 50 copies per milliliter or higher at week 48, determined with the use of the Food and Drug Administration snapshot algorithm.
Results
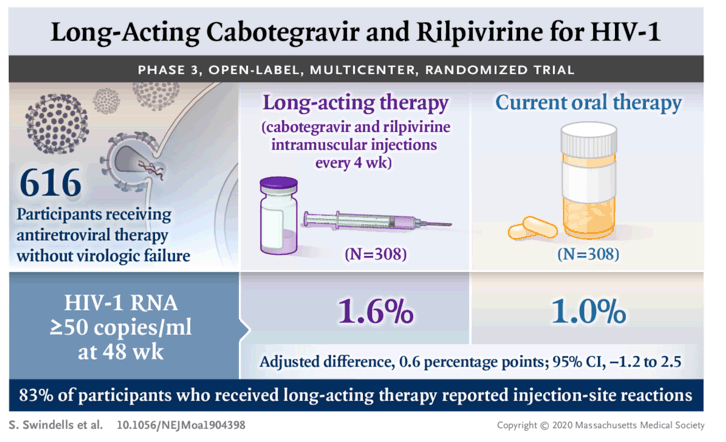
Treatment was initiated in 308 participants per group. At week 48, HIV-1 RNA levels of 50 copies per milliliter or higher were found in 5 participants (1.6%) receiving long-acting therapy and in 3 (1.0%) receiving oral therapy (adjusted difference, 0.6 percentage points; 95% confidence interval [CI], -1.2 to 2.5), a result that met the criterion for noninferiority for the primary end point (noninferiority margin, 6 percentage points). An HIV-1 RNA level of less than 50 copies per milliliter at week 48 was found in 92.5% of participants receiving long-acting therapy and in 95.5% of those receiving oral therapy (adjusted difference, -3.0 percentage points; 95% CI, -6.7 to 0.7), a result that met the criterion for noninferiority for this end point (noninferiority margin, -10 percentage points). Virologic failure was confirmed in 3 participants who received long-acting therapy and 4 participants who received oral therapy. Adverse events were more common in the long-acting–therapy group and included injection-site pain, which occurred in 231 recipients (75%) of long-acting therapy and was mild or moderate in most cases; 1% withdrew because of this event. Serious adverse events were reported in no more than 5% of participants in each group.
Conclusions
Monthly injections of long-acting cabotegravir and rilpivirine were noninferior to standard oral therapy for maintaining HIV-1 suppression. Injection-related adverse events were common but only infrequently led to medication withdrawal. (Funded by ViiV Healthcare and Janssen; ATLAS ClinicalTrials.gov number, NCT02951052. opens in new tab.)
Combination antiretroviral therapy for human immunodeficiency virus type 1 (HIV-1) infection provides durable viral suppression, which is associated with improved immunologic function and extended survival.1 Current guideline-recommended first-line regimens require lifelong daily oral therapy that can be burdensome, potentially affecting adherence and risking treatment failure.2-4 Surveys suggest that there is substantial interest among persons living with HIV for less frequent dosing options.5,6 Consequently, ongoing therapeutic research, including development of longer-acting injectable regimens, has been directed at simplifying antiretroviral therapy to improve satisfaction and facilitate adherence.6-8
Integrase strand-transfer inhibitors (INSTIs) or nonnucleoside reverse-transcriptase inhibitors (NNRTIs) are included in most guideline-recommended treatment regimens.1 Cabotegravir is structurally related to the approved INSTI dolutegravir and has a higher barrier to resistance than first-generation INSTIs.8,9 Rilpivirine is an approved second-generation NNRTI.10,11
Long-acting formulations of cabotegravir and rilpivirine can maintain exposure at plasma concentrations exceeding in vitro 90% inhibitory concentrations with monthly intramuscular injections.8 In LATTE-2 (Long-Acting Antiretroviral Treatment Enabling Trial 2), the percentage of participants with HIV-1 RNA suppression through 96 weeks was similar among those who switched to long-acting cabotegravir plus long-acting rilpivirine and those who continued oral cabotegravir-based therapy.12 Participants reported general satisfaction with injectable dosing and greater convenience than with previous oral therapy.13
Here we report the 48-week (primary end point) results of the phase 3 Antiretroviral Therapy as Long Acting Suppression (ATLAS) trial, the purpose of which was to establish whether switching to long-acting cabotegravir plus rilpivirine (long-acting therapy) is noninferior to continuation of current oral therapy among adults with virologically suppressed HIV-1 infection.
Results
Participants
Screening began on October 28, 2016, and the last participant completed week 48 on May 29, 2018. A total of 705 people were screened at 115 sites in 13 countries (see the Supplementary Appendix), and 618 underwent randomization (Figure 1B). In each treatment group, 308 participants started investigational treatment (intention-to-treat exposed population); 2 participants who had been assigned to the long-acting therapy regimen withdrew before starting treatment. Overall, the trial population was 33% female, 32% nonwhite, and a median of 42 years of age; 74% had CD4+ lymphocyte counts of 500 per cubic millimeter or higher (Table 1). The antiretroviral regimens at baseline included a two-NRTI backbone plus an NNRTI in 50% of participants, an INSTI in 33%, or a PI in 17% (Table S1 in the Supplementary Appendix). At trial entry, participants had been receiving their current antiretroviral regimen for a median of 4.3 years.
A total of 93% of participants completed maintenance-phase treatment through week 52, and 26 participants (8%) in the long-acting–therapy group and 18 (6%) in the oral-therapy group withdrew from the trial (Figure 1). Adverse events were the most frequent cause for withdrawal, occurring in 14 participants (5%) in the long-acting–therapy group and 5 (2%) in the oral-therapy group; 1% of participants in each group withdrew for lack of efficacy (Table 2 and Table 3). In the long-acting–therapy group, 98% of injections were administered within the permitted visit window (Fig. S1); 7 participants (2%) used oral bridging therapy (4 to 29 days) to cover missed or delayed injection visits.
Efficacy
In the intention-to-treat exposed population, HIV-1 RNA levels of 50 copies per milliliter or higher at week 48 were found in 5 participants (1.6%) in the long-acting–therapy group and 3 (1.0%) in the oral-therapy group (adjusted difference, 0.6 percentage points; 95% confidence interval [CI], -1.2 to 2.5); these results met the prespecified noninferiority criterion for the primary end point (Table 2). Similarly, the long-acting therapy was noninferior to oral therapy with respect to the key secondary end point of an HIV-1 RNA level of less than 50 copies per milliliter at week 48 (92.5% and 95.5%, respectively; adjusted difference, -3.0 percentage points; 95% CI, -6.7 to 0.7). No evidence of heterogeneity in these between-group differences was found across randomization strata or according to other baseline characteristics (Fig. S2). Results were consistent in the per-protocol population (Table 2), which excluded 30 participants for protocol deviations (Table S2). Results were also consistent with those of other secondary efficacy analyses (Table 2 and Table S3).
Resistance Analyses
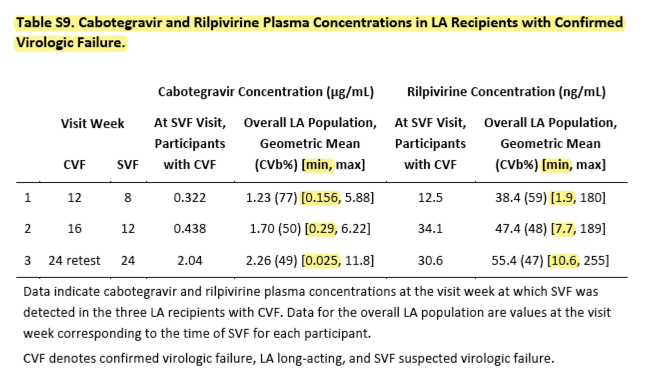
Three participants in the long-acting–therapy group had confirmed virologic failure — two with HIV-1 subtype A/A1 (failure occurred at week 8 in one participant and at week 20 in the other) and one with subtype AG (in whom failure occurred at week 12). Rilpivirine resistance–associated reverse-transcriptase mutations were detected in HIV-1 RNA samples from all three participants at the time of virologic failure, including E138A in one participant and E138K plus V108I in another (in both these participants, the same E138 mutations were present in HIV-1 DNA at baseline) and E138E/K plus the integrase mutation N155H in the third. These mutations reduced susceptibility to rilpivirine by a factor of 6.5, and cabotegravir susceptibility was reduced by a factor of 2.7 in the participant with N155H (Table S4). Two participants also had an L74I integrase polymorphism at baseline and at virologic failure, although this accessory mutation by itself does not decrease susceptibility to INSTIs.19 No participant with virologic failure missed an injection or received injections outside the permitted window. Four participants in the oral-therapy group had confirmed virologic failure, and reverse-transcriptase mutations were detected in three of these participants: one had the M184I mutation, one had M184V plus G190S, and one had M230M/I.
Safety and Side Effects
Adverse events were infrequent during the 4-week lead-in period with oral cabotegravir plus rilpivirine; three participants withdrew during this period. In the maintenance phase, 95% of participants in the long-acting–therapy group and 71% of participants in the oral-therapy group reported at least one adverse event (Table 3 and Table S5). Differences between the treatment groups were largely attributable to injection-site reactions, which occurred in 83% of participants in the long-acting–therapy group. Other drug-related adverse events, which were primarily of mild or moderate severity (grade 1 or 2), were more frequent with long-acting therapy (29%) than with oral therapy (3%). Grade 3 or 4 adverse events (primarily injection-site reactions) were more frequent in the long-acting–therapy group. Other than injection-site reactions, the most severe events reported as being related to the long-acting therapy were grade 3 pyrexia (one participant), nausea (one), diarrhea (one), and headache (two) and grade 4 lipase increase (one). At week 48, the median weight gains were 1.80 kg (interquartile range, -0.30 to 4.90) in the long-acting–therapy group and 0.30 kg (interquartile range, -1.60 to 2.50) in the oral-therapy group.
The incidence of serious adverse events was similar in the two groups (Table S6); one event (suicidal ideation in the oral-therapy group) was considered by the investigators to be related to the trial regimen. In each group, eight participants (3%) had disease progression to Centers for Disease Control and Prevention stage 3 or death (Table S3). Four participants in the long-acting–therapy group (1%) withdrew because of injection-site reactions; all four reported injection-site pain, and two also reported a nodule or swelling (Table S7). Other than injection-site reactions, no specific type of adverse event led to withdrawal in more than two participants in either group.
Among the participants who received long-acting therapy, 99% of injection-site reactions (Table 3) were of mild or moderate severity; no life-threatening or fatal (grade 4 or 5) reactions were reported, and 88% of reactions resolved within 7 days (median, 3 days). The most common injection-site reaction was pain (75% of participants); nodule (12%), induration (10%), and swelling (7%) were less common. Injection-site reactions were reported in 69% of participants after the initial 3-ml injections at week 4; frequencies of such reactions declined progressively after the subsequent 2-ml injections, reaching 11% at week 48 (Fig. S3).
Five participants in the long-acting–therapy group and one in the oral-therapy group had alanine aminotransferase elevations to at least 3 times the upper limit of the normal range (Table S8). Five of these events met protocol-defined liver-related stopping criteria. Among the participants who had these events, hepatitis A was diagnosed in three, hepatitis B in one, and hepatitis C in one. The trial treatment was stopped in all five participants and was restarted subsequently in one (in the oral-therapy group).
Pharmacokinetics
The concentrations of cabotegravir and rilpivirine in plasma during long-acting therapy were similar to those reported during oral therapy (Figure 2).20,21 Both drugs showed accumulation by a factor of approximately 2.3 from the first trough at week 8 to the trough at week 48, approximating steady-state drug concentrations. Geometric mean concentrations of cabotegravir and rilpivirine in plasma at week 48 (2.84 μg per milliliter and 90.3 ng per milliliter, respectively) were 17 times and 7.5 times as high as their respective protein-adjusted concentrations required for 90% viral inhibition, similar to outcomes after monthly dosing in a phase 2 study.12 All three participants in the long-acting–therapy group who had confirmed virologic failure received all injections on schedule; however, plasma concentrations of cabotegravir and rilpivirine at the time of failure were in the lower quartiles of the ranges of observed concentrations (Table S9).
Patient-Reported Outcomes
After 44 weeks, participants in the long-acting–therapy group reported substantially greater improvement from baseline in treatment satisfaction than participants in the oral-therapy group, as assessed with the HIVTSQs; the adjusted mean increase in score from baseline was 5.68 points higher (95% CI, 4.37 to 6.98) in the long-acting–therapy group than in the oral-therapy group (Table S10). This difference meets the threshold for the minimal clinically important difference according to the distribution-based approach. In a within-group comparison conducted at week 48 in the long-acting–therapy group, 97% of participants who responded to the questionnaire (266 of 273) and 86% of participants in the intention-to-treat exposed population (266 of 308) selected the injectable regimen over daily oral therapy as their preferred HIV treatment.
Discussion
Treatment simplification has been a focus of recent HIV-1 research to improve adherence, side effects, and quality of life. This trial shows successful treatment of HIV-1 infection with an all-injectable regimen as an alternative to daily oral treatment. Monthly dosing with longer-acting formulations of the INSTI cabotegravir and the NNRTI rilpivirine provided plasma concentrations of the drugs that were similar to those observed during daily oral therapy with cabotegravir and rilpivirine in combination with NRTIs.20,21 HIV-1 suppression through 48 weeks was maintained in similarly high percentages of participants with the injectable long-acting regimen and conventional three-drug oral regimens. In subgroup analyses, no meaningful differences in virologic outcomes were observed according to sex, third-agent class (INSTI, NNRTI, or PI) in previous oral regimens, or baseline disease or demographic characteristics.
Participants who received the long-acting therapy reported greater satisfaction and preferred the regimen over previous oral therapy. Although agreement to enroll in the trial implies willingness to try injectable therapy, most participants maintained a favorable view of the regimen even after 12 monthly injections.
Frequencies of serious adverse events were similar in the two treatment groups; no treatment-related serious adverse events were reported in the long-acting–therapy group. Injection-site reactions, primarily pain, were common after the first injection but became less frequent subsequently; 1% of participants discontinued long-acting therapy as a result of these events. As has occurred in previous switch studies, participants who switched to the long-acting therapy reported more adverse events than those who continued their familiar oral regimens, potentially contributing to the greater frequency of drug-related adverse events other than injection-site reactions in the long-acting–therapy group.22,23
All confirmed virologic failures in the long-acting therapy group occurred in participants with HIV-1 subtype A or AG, a finding that warrants further investigation. Low trough concentrations of the trial drugs may have contributed to virologic failure in recipients of long-acting therapy; however, all injections in participants with virologic failure were received within the prescribed visit window, and a clear relationship between drug concentrations and infrequent virologic failure could not be established.
This trial has several limitations. The trial population had stably suppressed HIV-1 infection and substantial treatment histories but no previous virologic failure, which may limit generalizability. In this regard, the FLAIR trial is evaluating patients who had not previously received antiretroviral therapy and who switched to the long-acting regimen after having viral suppression with oral dolutegravir–abacavir–lamivudine, and the ongoing ATLAS-2M trial is comparing long-acting treatment intervals of 4 and 8 weeks. Both trials include extension phases to evaluate longer-term outcomes. Studies involving populations that may derive benefit from the long-acting regimen, such as patients with adherence challenges or gastrointestinal absorption issues, would provide additional useful information.
In this trial, the monthly injectable long-acting regimen was noninferior to standard once-daily oral therapy for maintaining HIV-1 suppression. Injection-site reactions were common but generally were of mild or moderate severity and transient, and participant satisfaction was higher with the injectable regimen. This regimen may provide a new treatment option for patients living with HIV.
|
|
| |
| |
|
|
|