| |
Long-acting implants to treat and prevent HIV infection
|
| |
| |
Download the PDF here
Download the PDF here
Abstract
Purpose of review
Subcutaneous implants are a promising technology to enable long-acting parenteral delivery of antiretroviral drugs (ARV) because they may be able to provide protective drugs concentrations for a year or longer following a single implant. The present review covers the current status of preclinical and clinical development of antiretroviral implants.
Recent findings
Over the past three decades, subcutaneous implants have been widely used for long-acting hormonal contraception and the treatment of hormonally-driven malignancies. They are economical and scalable to manufacture, but require special procedures for insertion and removal. They are generally well tolerated, and can remain in place for up to five years. As long-acting delivery of ARV would confer significant advantages, a few investigational implants are under development for the delivery of ARV; most remain at preclinical stages of development. Islatravir, a potent nucleoside analog reverse transcriptase translocation inhibitor that shows particular promise, has entered clinical testing in implant form. Investigational implants containing tenofovir alafenamide and nevirapine, and entecavir (for hepatitis B virus) have been developed and tested in animal models, with varying degrees of success. There is also burgeoning interest in bioerodable implant formulations of established ARVs.
Summary
LARV implants are a promising new technology, but are in early stages of clinical development. Their potential advantages include more consistent and predictable drug release than that provided by intramuscular injections, the possibility of combining several partner drugs into one implant, and the fact that implants can be removed in the case of a desire to stop treatment or the development of adverse events.
LONG-ACTING ANTIRETROVIRAL IMPLANTS IN PRECLINICAL AND CLINICAL DEVELOPMENT
ARV agents that are most suitable for implant formulation and delivery are those with exceptionally high antiviral potency. Because the allowable mass dose of active pharmaceutical ingredient that can loaded into a single implant rod is small, the agent or agents involved must have extraordinary capacity to inhibit viral replication at low concentrations, otherwise drug delivery by implant becomes infeasible even if one considers inserting multiple rods at the same time. Transdermal strategies (such as drug delivery via matrix-based adhesive patch or reservoir-based patches) generally depend on the drug having adequate properties for passively permeating through skin—these include low molecular weight of less than 500 Da, melting point less than 250°C, and moderate log P[1,2,3 ,5 ].
In contrast to most ARVs, hormones used in contraception are exceedingly potent. For perspective, a single 3-year etonogestrel (Nexplanon) implant delivers an average daily dose of etonogestrel of only 62 μg (0.06 mg) per day [6], and a single levonorgestrel (Jadelle) 75 mg implant delivers an expected daily dose of 82 μg (0.08 mg) over the 5-year insertion period [7]. In the case of the levonorgestrel implant, the release rate is known to slow over the first 2 years after insertion, dropping from approximately 100 μg/day during the first month to 40 μg/day at 12 months and 30 μg/day after month 24 [7] (see Table 1 for more details). Few marketed drugs have a daily dose of active pharmaceutical ingredient this low. The most potent approved antiretroviral drugs, tenofovir alafenamide and rilpivirine, have daily oral doses of 25 mg/day.
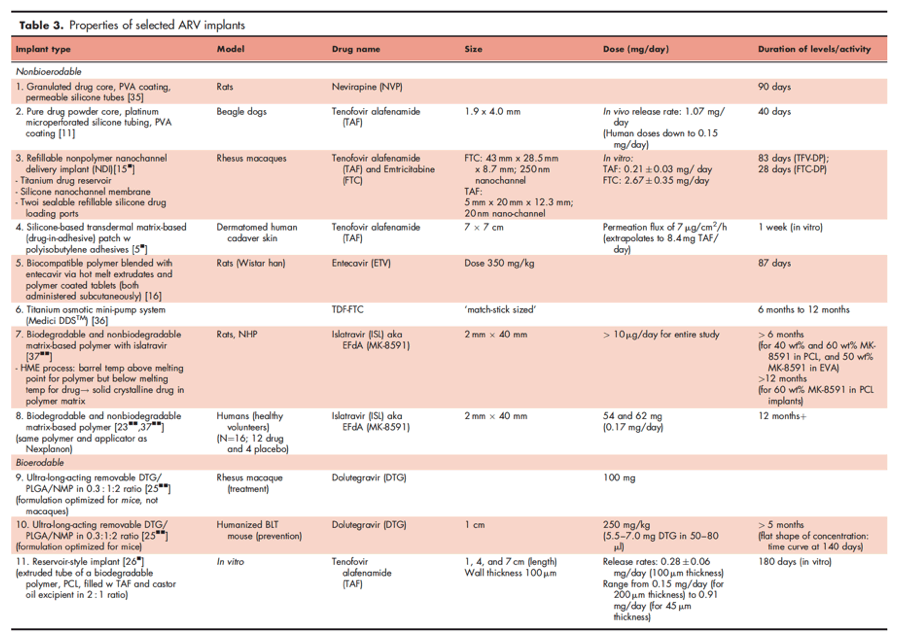
Several antiretroviral compounds have now been identified whose in vitro potency approaches that seen with drugs used in implants marketed for other indications. These are reviewed below, and a summary of candidate ARV implants is provided in Table 3.
Tenofovir prodrugs
Tenofovir is the world's most widely used antiretroviral, and is currently marketed as two different oral prodrugs. Since its intracellular active metabolite, tenofovir-diphosphate (TFV-diphosphate), has an estimated half-life of 60-100 h, tenofovir prodrugs could easily be given less frequently than once per day while maintaining antiviral effects [8]. The prodrug tenofovir alafenamide (TAF) is 10 times more potent than tenofovir disoproxil fumarate (TDF), and has the added advantage that it is taken up into cells from the plasma as the prodrug, which is converted to the parent drug and then to the active intracellular diphosphate inside the cell [9]. In addition, from the perspective of prevention, TAF has activity against both hepatitis B virus (HBV) and HIV, and could potentially prevent both infections, in addition to treating them in those who have already acquired coinfection [10].
Several approaches have been pursued for the creation of a TAF implant. In the first, investigators loaded pure TAF powder into platinum cured microperforated silicone tubing that was coated with polyvinyl alcohol (PVA). The 1.9 x 40 mm implants were inserted subcutaneously into beagle dogs. This implant produced measurable plasma concentrations of tenofovir for more than 6 weeks and delivered tenofovir at an approximately constant rate for 5 to 40 days after implantation [11]. The zero-order, linear kinetics were observed over the first 30 days, after which there was a steeper decline with first-order kinetics because of drug depletion. The implants were well tolerated in the beagles, without significant AEs. Strikingly, intracellular tenofovir-diphosphate concentrations were 11-32 times higher than those associated with effective preexposure prophylaxis in humans in the iPrEX study of 16-48 fmol/106 cells after using TDF [11,12]. One advantage of this implant technology is that its physical characteristics (tube diameter, number, and size of perforations) can be easily modified to alter the release rate of drug. This device has now entered Phase I testing in humans, although no data are yet publically available.
Another approach for innovative delivery of TAF is a silicone-based transdermal matrix-based (drug-in-adhesive) patch with polyisobutylene adhesives, which has been tested on dermatomed human cadaver skin and has been established to yield a permeation flux of 7 μg/cm2/h over 1 week, which extrapolates to a release rate of 8.4 mg of TAF per day. This noninvasive approach offers convenience, simplicity, has no procedural requirements and thus stands to be more user friendly [5 ].
The combination of TAF and FTC (shown to be efficacious as PrEP in the DISCOVER trial) [13,14] has been investigated in rhesus macaques, using a refillable nonpolymer nanofluidic implant (nanochannel delivery implant, NDI) consisting of a titanium drug reservoir, silicone nanochannel membrane, and two sealable refillable silicone drug loading ports [15 ]. The advantage of this approach is that when drug is exhausted, more drug can be administered transcutaneously into the ports, without the need to remove and reimplant a new device. The NDI achieved concentrations of TFV-diphosphate in PBMCs that reached target 3 days after implantation, and continued to increase from a mean of 71.7 ± 29.7 to 533.3 ± 198.4 fmol/106 PBMCs over 70 days (equivalent to 12 times the iPrEX EC90) [12,15 ]. Concentrations of FTC-TP in PBMCs were detected and sustained from day 3 to day 28 (1223.7 ± 416.8 to 1656.7 ± 290.8 fmol/106 PBMCs), but unlike TAF did not attain target protective concentrations (equivalent to an EC90 extrapolated from the iPrEX study of 5000-6000 fmol/106 freshly lysed PBMCs). The NDI was fairly well tolerated, but 1 in 3 animals had swelling/seroma around the device, which resolved in 7 days, one animal had dehiscence over the NDI at 60 days, and 2 animals developed skin ulceration over the implant at day 70.
4'-Ethynyl-2-fluoro-2'-deoxyadenosine (MK8591; islatravir)
4'-Ethynyl-2-fluoro-2'-deoxyadenosine (EFdA; MK8591; islatravir [ISV]) is a highly potent nucleoside reverse transcriptase inhibitor with a novel mechanism of action [17]. ISV is a 4'-substituted NRTI that retains its 3'-hydroxyl group, unlike any other currently approved NRTI. As a consequence, this drug has higher affinity for the active site of the HIV reverse transciptase, contributing to its potency. The incorporation of ISV-monophosphate into the RT active site blocks primer translocation and halts HIV replication without causing chain termination, since the drug contains a 3'-hydroxyl. ISV also has a halogen substitution at the 2-position of the adenine ring that impairs degradation by adenosine deaminase and contributes to the prolonged intracellular half-life of active phosphorylated metabolite ISV-TP, estimated at greater than 72 h [18]. ISV has substantial efficacy in treatment and prevention of HIV in animal models with doses as low as 0.1 mg/kg/day. In animals, the drug is safe and well tolerated across a range of doses [19].
ISV has a favorable resistance profile for HIV treatment and prevention. The major resistance mutation associated with the use of this drug has been M184V, which only modestly reduces drug sensitivity in vitro[20]. These suggest the possibility of using ISV in patients already harboring M184V-containing HIV, and of using this drug in combination with other existing antiretroviral NRTIs and other drug classes. ISV is approximately 10-fold more potent against HIV-2 isolates than against HIV-1 in vitro, indicating broad-spectrum activity and possible global utility for this agent [21]. Possible uses for ISV implants, like those for TAF, include preexposure prophylaxis as a single agent, or eventual combination with other long-acting antiretroviral drugs for HIV treatment.
In studies comparing two nondegradable polymer implants in rats, the apparent ISV plasma half-life was nearly 100 days, suggesting the possibility for developing human implants with a dosing interval of 1 year or longer [22]. Phase I clinical studies of ISV are currently underway in human subjects, and preliminary findings in healthy human volunteers showed that a 62 mg implant can achieve a constant, linear (zero-order) release of drug that results in levels of triphosphorylated active metabolite [ISV-TP] in PBMCs of 0.076 pmol/106 over 12 months, which is approximately 7 times the IC50in vitro for wild-type virus, maintaining intracellular concentrations above the target of 0.05 pmol/106 cells [23 ]. The t1/2 observed in humans of parent ISV was 50-60 h and of active metabolite ISV-TP was 120-177 h. The implants were generally well tolerated, though with local erythema, itching, and induration that was mild and to some extent dose-dependent. Importantly, based on modeling, the projected time at which concentrations fall below the target is not until 16 months after implant was placed. This raises the possibility of implant placement frequency less often than yearly for HIV treatment and prevention [17].
Bioerodable implants
Some problems associated with medical implants could be addressed with newer technologies. For example, bioerodable/biodegradable implants, for instance using polylactic acid or polylactide, have been deployed in drug-eluting stents [24]. In the case of a bioerodadable antiretroviral 'implant,' the formulation would be injected, forming a solid gel that would disperse over several months' time. This has the advantage that the implant system does not require eventual removal. However, if there is the desire to remove the implant for adverse drug effects, surgical removal becomes more difficult, and potentially impossible, several months after injection, because the implant has dispersed.
One example of a bioerodable implant currently in preclinical testing is ultra-long-acting dolutegravir (DTG). Ultra-long-acting DTG delivered via a removable implant system has been tested in a macaque model for HIV treatment and a humanized bone marrow-liver-thymus (BLT) mouse for HIV prevention [25 ]. The technology involves subcutaneous injection of an admixture of DTG, PLGA, and NMP in a ratio of 0.3:1:2. Liquid drug is injected and then solidifies over the first 48 h in vivo into an implant once it reaches the aqueous environment of the body, and then biodegrades slowly, resulting in sustained drug release. This delivers drug at effective concentrations for up to 9 months and can be safely removed should an adverse event or pregnancy occur. Implants were well tolerated in both animal models, with no local necrosis. The sustained drug concentrations resulted in both suppression of viremia in the macaques and prevention from acquiring HIV via a high-dose repeat vaginal challenge.
Another bioerodable approach that has been tested in vitro involves the creation of an extruded tube made of a biodegradable polymer, PCL of various thicknesses, filled with TAF and castor oil excipient in a 2 : 1 ratio [26 ,27]. The implant achieved over 180 days daily TAF release rates of 0.28 ± 0.06 mg/day (100 μm thickness), with a range from 0.15 mg/day (for 200 μm thickness) to 0.91 mg/day (for 45 μm thickness). This is above the concentration of 0.15 mg/day that is estimated to be sufficient to achieve target TFV-diphosphate intracellular concentrations for anti-HIV efficacy and protection. Daily release rates can be modified by changing the implant surface area, PCL tube wall thickness (the thinner the more drug release), or PCL crystallinity.
ANTIRETROVIRAL IMPLANTS: CHALLENGES AND OPPORTUNITIES
Theoretical advantages of implant technology for HIV treatment and prevention include the ability to remove the device in the case of side effects or the desire to end therapy, less frequent dosing because of the slow release of drug and longer apparent half-life, a lower drug dose per day because of potency and formulation properties, protection from poor adherence as compared to daily oral drugs, and the possibility of directly observed therapy. In the case of formulations with a prolonged low-level pharmacokinetic 'tail,' the device can simply be removed, allowing drug concentrations to decline rapidly. Medical implants are placed where they cannot generally be detected by partners, family members, or colleagues, affording better protection of health privacy. The widespread use of contraceptive implants in low income countries proves that these devices can be implemented cheaply and safely in remote areas.
Although implants have some clear advantages over injectable formulations, they also have some drawbacks (see Table 2). This includes the need for insertion and removal by well trained personnel using sterile technique. Because these devices are removable, it is likely that a novel antiretroviral developed for implant technology would not require a partnered oral formulation or oral lead-in to rule out toxicity or hypersensitivity, as is the case for injectable cabotegravir and rilpivirine [28,29]. Nonetheless, different dose formulations would still need to be developed to allow for dose escalation in clinical dose-finding and safety studies.
In HIV prevention applications, an antiretroviral implant could be developed as a stand-alone. However, partner formulations would need to be identified for HIV treatment. Ideally, a single implant could contain two or more antiretrovirals with similar pharmacokinetic properties, or two separate implants could be inserted simultaneously. As an intermediate approach, a long-lasting implant could be paired with periodic injections of an intramuscular formulation like LA-cabotegravir or LA-rilpivirine, or one or more long-acting, broadly neutralizing monoclonal antibodies [30,31].
CONCLUSION
Implants show great promise for HIV treatment and prevention. Potential advantages over intramuscular injections include the fact that implants are removable in cases of toxicity, produce more consistent and predictable drug release, have pharmacokinetic properties that may not be dependent on the injection site, and likely will not require an oral lead-in phase. In addition, implants can sustain therapeutic drug concentrations and remain in place for years, based on experience with hormonal contraception. However, implants require specialized training for insertion and removal, a minor surgical procedure if they need to be removed, and are more expensive and complex to manufacture than injectables, making transition to a generic marketplace more difficult as compared to injectable formulations. Many of these problems can and will be solved with future innovation, adding to the number of antiretroviral choices available to people living with or at risk of HIV.
------------------------------------------
Design and Testing of a Cabotegravir Implant for HIV Prevention
See pdf attached
Abstract
Long-acting antiretroviral implants could help protect high-risk individuals from HIV infection. We describe the design and testing of a long-acting reservoir subcutaneous implant capable of releasing cabotegravir for several months. We compressed cabotegravir and excipients into cylindrical pellets and heat-sealed them in tubing composed of hydrophilic poly(ether-urethane) -. The implants have a 47 mm lumen length, 3.6 mm outer diameter, and 200 μm wall thickness. Four cabotegravir pellets were sealed in the membrane, with a total drug loading of 274 ± 3 mg. In vivo, the implants released 348 ± 107 μg/day (median value per implant, N = 41) of cabotegravir in rhesus macaques. Five implants generated an average cabotegravir plasma concentration of 373 ng/ml in rhesus macaques. The non-human primates tolerated the implant without gross pathology or microscopic signs of histopathology compared to placebo implants. Cabotegravir plasma levels in macaques dropped below detectable levels within two weeks after the removal of the implants.
Conclusion
We describe a subcutaneous reservoir implant capable of delivering approximately 350 μg/day of cabotegravir over three months in rhesus macaques. In 90 days we observed implant B released 11% of the total cabotegravir load suggesting these implants could deliver cabotegravir for a year in the nonhuman primate model. All components used in this implant have a history of previous human use in parenteral products. The CAB drug loading is high (∼87% by weight), allowing efficient use of the implant volume. The methods described are at present capable of reproducibly manufacturing batch sizes up to 1000 implants with common pharmaceutical operations (hot melt extrusion for the RCM tubing and tablet compression for the core pellet) and an impulse sealer. Additionally, this reservoir system design is more complicated and likely more expensive than simple matrix extrusion dosage forms. The cabotegravir release rate is adjustable through the modulation of the geometry and composition of the HPEU HPEU RCM. While the implants generally give reproducible release profiles, disproportionation and counter ion-exchange likely affect transport in complex ways that require further study. Other methods to increase core drug concentration and release rate, like the use of cyclodextrins [25] or high molecular weight pH modifiers , or improved polymers with higher drug diffusivity and solubility may allow improvement in the release rate of cabotegravir from this type of reservior implant.
|
|
| |
| |
|
|
|