| |
FDA Approves New Treatment Baraclude (entecavir) for Chronic Hepatitis B
|
| |
| |
Following this brief announcement from the FDA is selected information from the Baraclude Label:
--Results from Clinical Studies (HIV-negative & HIV-positive studies)
--Exacerbations of Hepatitis After Discontinuation of Treatment
--adverse reactions
--dosing in renal impairment
-- Carcinogenesis, Mutagenesis, Impairment of Fertility
--resistance, cross-resistance
--mechanism of action
--in vitro antiviral activity
-- Pregnancy Category C
--use in racial/ethnic groups
--drug interactions
FDA Hearing Report
http://www.natap.org/2005/HBV/031505_02.htm
The Food and Drug Administration (FDA) announced the approval of Baraclude (entecavir) tablets and oral solution for the treatment of chronic hepatitis B in adults.
Chronic hepatitis B is a serious disease caused by the hepatitis B virus (HBV) that attacks the liver. The virus can cause lifelong infection, cirrhosis (scarring) of the liver, liver cancer, liver failure, and death. According to the Centers for Disease Control and Prevention, approximately 1.25 million Americans are chronically infected with the HBV virus.
Baraclude slows the progression of chronic hepatitis B by interfering with viral reproduction.
FDA based its approval of Baraclude on the results of three studies in which Baraclude was compared to another anti-viral drug, lamivudine.
In all three clinical studies, patients treated with Baraclude showed significant improvement in the liver inflammation caused by HBV and an improvement in the degree of liver fibrosis (scarring). In addition, a higher percentage of patients treated with Baraclude showed significant improvement compared to lamivudine.
The major adverse events associated with the use of Baraclude were of the type typically seen with HBV therapy. They include severe, acute exacerbation of hepatitis B after discontinuation of Baraclude, headache, abdominal pain, diarrhea, fatigue, and dizziness. The labeling for Baraclude states that patients who discontinue Baraclude should be monitored at repeated intervals over a period of time for liver function.
Baraclude's sponsor, Bristol-Myers Squibb Company of Wallingford, Conn., has committed to conducting a large post-marketing study of Baraclude (entecavir) to evaluate the risks of cancers and liver related complications.
The FDA approved Baraclude (entecavir) for the treatment of chronic hepatitis B virus infection in adults with evidence of active viral replication, and either evidence of persistent elevations in serum aminotransferases (ALT or AST), or histologically active disease.
This indication is based on histologic, virologic, biochemical, and serologic responses after one year of treatment in nucleoside-treatment-naive and lamivudine-resistant adult patients with HBeAg*-positive, or HBeAg-negative chronic HBV infection with compensated liver disease, and on more limited data in adult patients with HIV/HBV co-infection who have received prior lamivudine therapy. *(hepatitis B e antigen)
Limited data about Baraclude in patients with HIV/HBV co-infection who received prior lamivudine therapy are presented in the label. Please refer to the attached label and the Special Population section under Description of Clinical Studies for information on HIV/HBV co-infected patients.
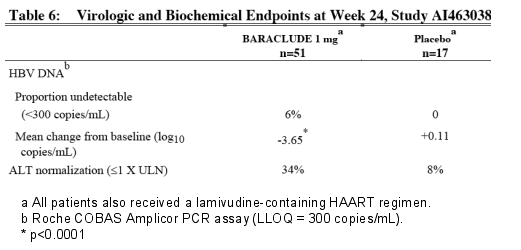
In summary, Study AI463038 was a randomized, double-blind, placebo-controlled study of Baraclude versus placebo in 68 patients co-infected with HIV and HBV who experienced recurrence of HBV viremia while receiving a lamivudine-containing highly active antiretroviral therapy (HAART) regimen. Patients continued their lamivudine-containing HAART regimen (lamivudine dose 300 mg/day) and were assigned to add either BARACLUDE 1 mg once daily (51 patients) or placebo (17 patients) for 24 weeks followed by an open-label (non-blinded) phase for an additional 24 weeks where all patients received BARACLUDE.
At baseline, patients had a mean serum HBV DNA level by PCR of 9.13 log10 copies/mL. Ninety-nine percent of patients were HBeAg-positive at baseline, with a mean baseline ALT level of 71.5 U/L. Median HIV RNA level remained stable at approximately 2 log10 copies/mL through 24 weeks of blinded therapy.
The proportion of HIV/HBV co-infected patients with HBV DNA < 300 copies/mL was 6% for the BARACLUDE 1 mg group versus 0% for the placebo group. The mean change from baseline for HBV DNA was -3.65 log10 copies/mL for the BARACLUDE 1 mg group versus +0.11 log10 copies/mL for the placebo group. Thirty-four percent of patients in the Baraclude 1 mg group had ALT normalization (< 1 x ULN) compared to 8% of patients in the placebo group.
There are no data for patients with HIV/HBV co-infection who have not received prior lamivudine therapy.
Richard Klein
Office of Special Health Issues
Food and Drug Administration
Kimberly Struble
Division of Antiviral Drug Products
Food and Drug Administration
From The Baraclude Label
The recommended dose of BARACLUDE for chronic hepatitis B virus infection in nucleoside-treatment-naive adults and adolescents 16 years of age and older is 0.5 mg once daily.
The recommended dose of BARACLUDE in adults and adolescents (≥16 years of age) with a history of hepatitis B viremia while receiving lamivudine or known lamivudine resistance mutations is 1 mg once daily.
BARACLUDE should be administered on an empty stomach (at least 2 hours after a meal and 2 hours before the next meal).
RESISTANCE
In Vitro
In cell-based assays, 8- to 30-fold reductions in entecavir phenotypic susceptibility were observed for lamivudine-resistant strains. Further reductions (>70-fold) in entecavir phenotypic susceptibility required the presence of primary lamivudine resistance amino acid substitutions (rtL180M and/or rtM204V/I) along with additional substitutions at residues rtT184, rtS202, or rtM250, or a combination of these substitutions with or without an rtI169 substitution in the HBV polymerase.
Clinical Studies
Nucleoside-naive patients: Eighty-one percent of HBV chronically infected nucleoside-naive patients receiving entecavir 0.5 mg once daily achieved a reduction in viral load to <300 copies/mL at 48 weeks. Genotypic analysis of serum HBV DNA from nucleoside-naive HBeAg-positive (Study AI463022; n=219) or HBeAg-negative (Study AI463027; n=211) patients detected no genotypic changes in the HBV polymerase associated with phenotypic resistance to entecavir at Week 48. No genotypic or phenotypic evidence of entecavir resistance was detected in the 2 patients who experienced a confirmed virologic rebound (≥1 log increase from nadir) in Study AI463022. Lamivudine-refractory patients: Twenty-two percent of lamivudine-refractory patients with chronic HBV infection achieved HBV DNA levels <300 copies/mL at Week 48 on entecavir 1 mg once daily. Genotypic analysis of clinical samples from those patients with detectable viral DNA identified 7% (13/189) with evidence of emerging entecavir resistance-associated substitutions at rtI169, rtT184, rtS202, and/or rtM250 by Week 48 when pre-existing lamivudine resistance mutations rtL180M and/or rtM204V/I were present. Of the 13 patients with genotypic resistance, 3 experienced virologic rebound (≥1 log increase from nadir) by Week 48, with the majority of these 13 patients experiencing virologic rebound beyond Week 48.
Cross-resistance
Cross-resistance has been observed among HBV nucleoside analogues. In cell-based assays, entecavir had 8- to 30-fold less inhibition of replication of HBV containing lamivudine resistance mutations rtL180M and/or rtM204V/I than of wild-type virus. Recombinant HBV genomes encoding adefovir resistance-associated substitutions at either rtN236T or rtA181V remained susceptible to entecavir. HBV isolates from lamivudine-refractory patients failing entecavir therapy were susceptible in vitro to adefovir but retained resistance to lamivudine.
Description of Clinical Studies (HIV-negative & HIV-positive studies)
The safety and efficacy of BARACLUDE were evaluated in three Phase 3 active-controlled trials. These studies included 1633 patients 16 years of age or older with chronic hepatitis B infection (serum HBsAg-positive for at least 6 months) accompanied by evidence of viral replication (detectable serum HBV DNA, as measured by the bDNA hybridization or PCR assay). Patients had persistently elevated ALT levels ≥1.3 times the upper limit of normal (ULN) and chronic inflammation on liver biopsy compatible with a diagnosis of chronic viral hepatitis. The safety and efficacy of BARACLUDE were also evaluated in a study of 68 patients co-infected with HBV and HIV.
Nucleoside-Naive Patients With Compensated Liver Disease
HBeAg-positive:
Study AI463022 was a multinational, randomized, double-blind study of BARACLUDE 0.5 mg once daily versus lamivudine 100 mg once daily for 52 weeks in 709 (of 715 randomized) nucleoside-naive patients with chronic hepatitis B infection and detectable HBeAg. The mean age of patients was 35 years, 75% were male, 57% were Asian, 40% were Caucasian, and 13% had previously received interferon-_. At baseline, patients had a mean Knodell Necroinflammatory Score of 7.8, mean serum HBV DNA as measured by Roche COBAS Amplicor¨ PCR assay was 9.66 log10 copies/mL, and mean serum ALT was 143 U/L. Paired, adequate liver biopsy samples were available for 89% of patients.
HBeAg-negative (anti-HBe positive/HBV DNA positive):
Study AI463027 was a multinational, randomized, double-blind study of BARACLUDE 0.5 mg once daily versus lamivudine 100 mg once daily for 52 weeks in 638 (of 648 randomized) nucleoside-naive patients with HBeAg-negative (HBeAb-positive) chronic hepatitis B infection. The mean age of patients was 44 years, 76% were male, 39% were Asian, 58% were Caucasian, and 13% had previously received interferon-_. At baseline, patients had a mean Knodell Necroinflammatory Score of 7.8, mean serum HBV DNA as measured by Roche COBAS Amplicor PCR assay was 7.58 log10 copies/mL, and mean serum ALT level was 142 U/L. Paired, adequate liver biopsy samples were available for 88% of patients.
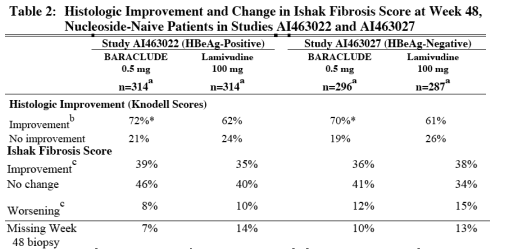
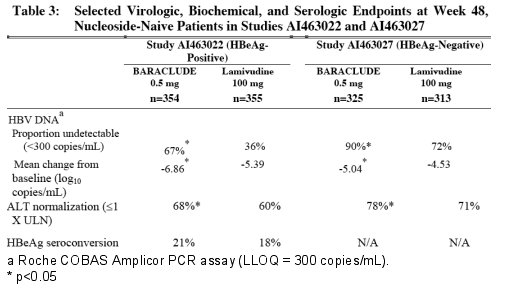
In Studies AI463022 and AI463027, BARACLUDE was superior to lamivudine on the primary efficacy endpoint of Histologic Improvement, defined as ≥2-point reduction in Knodell Necroinflammatory Score with no worsening in Knodell Fibrosis Score at Week 48, and on the secondary efficacy measures of reduction in viral load and ALT normalization. Histologic Improvement and change in Ishak Fibrosis Score are shown in Table 2. Selected virologic, biochemical, and serologic outcome measures are shown in Table 3
|
|
| |
 |
| |
a Patients with evaluable baseline histology (baseline Knodell Necroinflammatory Score ≥2).
b ≥2-point decrease in Knodell Necroinflammatory Score from baseline with no worsening of the Knodell Fibrosis Score.
c For Ishak Fibrosis Score, improvement = ≥1-point decrease from baseline and worsening = ≥1-point increase from baseline.
* p<0.05
Histologic Improvement was independent of baseline levels of HBV DNA or ALT.
|
|
| |
 |
| |
Lamivudine-Refractory Patients
Study AI463026 was a multinational, randomized, double-blind study of BARACLUDE in 286 (of 293 randomized) patients with lamivudine-refractory chronic hepatitis B infection. Patients receiving lamivudine at study entry either switched to BARACLUDE 1 mg once daily (with neither a washout nor an overlap period) or continued on lamivudine 100 mg for 52 weeks. The mean age of patients was 39 years, 76% were male, 37% were Asian, 62% were Caucasian, and 52% had previously received interferon-_. The mean duration of prior lamivudine therapy was 2.7 years, and 85% had lamivudine resistance mutations at baseline by an investigational line probe assay. At baseline, patients had a mean Knodell Necroinflammatory Score of 6.5, mean serum HBV DNA as measured by Roche COBAS Amplicor PCR assay was 9.36 log10 copies/mL, and mean serum ALT level was 128 U/L. Paired, adequate liver biopsy samples were available for 87% of patients.
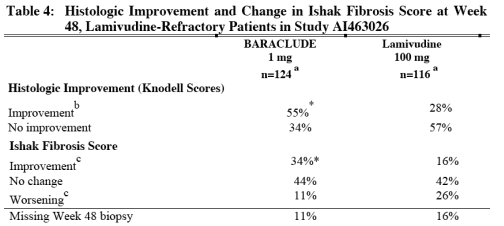
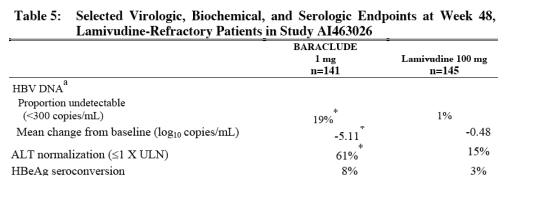
BARACLUDE was superior to lamivudine on the coprimary endpoint of Histologic Improvement (using the Knodell Score at Week 48). These results and change in Ishak Fibrosis Score are shown in Table 4. Table 5 shows selected virologic, biochemical, and serologic endpoints.
|
|
| |
 |
| |
a Patients with evaluable baseline histology (baseline Knodell Necroinflammatory Score ≥2).
b ≥2-point decrease in Knodell Necroinflammatory Score from baseline with no worsening of the Knodell Fibrosis Score.
c For Ishak Fibrosis Score, improvement = ≥1-point decrease from baseline and worsening = ≥1-point increase from baseline.
* p<0.01
Histologic Improvement was independent of baseline levels of HBV DNA or ALT.
|
|
| |
 |
| |
Post-Treatment Follow-up
The optimal duration of therapy with BARACLUDE is unknown. According to protocol-mandated criteria in the Phase 3 clinical trials, patients discontinued BARACLUDE or lamivudine treatment after 52 weeks according to a definition of response based on HBV virologic suppression (<0.7 MEq/mL by bDNA assay) and loss of HBeAg (in HBeAg-positive patients) or ALT normalization (<1.25 X ULN, in HBeAg-negative patients) at Week 48. For the 21% of nucleoside-naive, HBeAg-positive BARACLUDE-treated patients who met response criteria, response was sustained throughout the 24-week post-treatment follow-up period in 82%. For the 85% of nucleoside-naive, HBeAg-negative BARACLUDE-treated patients who met response criteria, response was sustained throughout the 24-week post-treatment follow-up period in 48%. Few lamivudine-refractory patients met the response criteria and were eligible to discontinue treatment. These protocol-specified patient management guidelines are not intended as guidance for clinical practice.
HIV-HBV Positive Study
Study AI463038 was a randomized, double-blind, placebo-controlled study of BARACLUDE versus placebo in 68 patients co-infected with HIV and HBV who experienced recurrence of HBV viremia while receiving a lamivudine-containing highly active antiretroviral (HAART) regimen. Patients continued their lamivudine-containing HAART regimen (lamivudine dose 300 mg/day) and were assigned to add either BARACLUDE 1 mg once daily (51 patients) or placebo (17 patients) for 24 weeks followed by an open-label phase for an additional 24 weeks where all patients received BARACLUDE. At baseline, patients had a mean serum HBV DNA level by PCR of 9.13 log10 copies/mL. Ninety-nine percent of patients were HBeAg-positive at baseline, with a mean baseline ALT level of 71.5 U/L. Median HIV RNA level remained stable at approximately 2 log10 copies/mL through 24 weeks of blinded therapy. Virologic and biochemical endpoints at Week 24 are shown in Table 6. There are no data in patients with HIV/HBV co-infection who have not received prior lamivudine therapy.
|
|
| |
 |
| |
Exacerbations of Hepatitis After Discontinuation of Treatment
Severe acute exacerbations of hepatitis B have been reported in patients who have discontinued anti-hepatitis B therapy, including entecavir. Hepatic function should be monitored closely with both clinical and laboratory follow-up for at least several months in patients who discontinue anti-hepatitis B therapy. If appropriate, initiation of anti-hepatitis B therapy may be warranted
Exacerbations of Hepatitis After Discontinuation of Treatment
In the Phase 3 studies, a subset of patients was allowed to discontinue treatment at 52 weeks if they achieved a protocol-defined response to therapy. An exacerbation of hepatitis or ALT flare was defined as ALT >10 X ULN and >2 X the patientÕs baseline level. As demonstrated in Table 9, a proportion of patients in the nucleoside-naive studies experienced post-treatment ALT flares. The number of lamivudine-refractory patients eligible to discontinue treatment was small, and rates of post- treatment flares in this population could not be determined. If BARACLUDE is discontinued without regard to treatment response, the rate of post-treatment flares could be higher.
|
|
| |
 |
| |
a Median time to off-treatment exacerbation was 23 weeks for BARACLUDE-treated patients and 12 weeks for lamivudine-treated patients.
b Median time to off-treatment exacerbation was 24 weeks for BARACLUDE-treated patients and 9 weeks for lamivudine-treated patients.
ADVERSE REACTIONS
Assessment of adverse reactions is based on four studies (AI463014, AI463022, AI463026, and AI463027) in which 1720 patients with chronic hepatitis B infection received double-blind treatment with BARACLUDE 0.5 mg/day (n=679), BARACLUDE 1 mg/day (n=183), or lamivudine (n=858) for up to 107 weeks. Median duration of therapy was 54 weeks for BARACLUDE-treated patients and 53 weeks for lamivudine-treated patients in Studies AI463022 and AI463027 and 69 weeks for BARACLUDE-treated patients and 52 weeks for lamivudine-treated patients in Studies AI463026 and AI463014. The safety profiles of BARACLUDE and lamivudine were comparable in these studies. The safety profile of BARACLUDE 1 mg (n=51) in HIV/HBV co-infected patients enrolled in Study AI463038 was similar to that of placebo (n=17) through 24 weeks of blinded treatment and similar to that seen in non-HIV infected patients.
The most common adverse events of any severity with at least a possible relation to study drug for BARACLUDE-treated patients were headache, fatigue, dizziness, and nausea. The most common adverse events among lamivudine-treated patients were headache, fatigue, and dizziness. One percent of BARACLUDE-treated patients in these four studies compared with 4% of lamivudine-treated patients discontinued for adverse events or abnormal laboratory test results.
Drug Interactions
Since entecavir is primarily eliminated by the kidneys coadministration of BARACLUDE with drugs that reduce renal function or compete for active tubular secretion may increase serum concentrations of either entecavir or the coadministered drug. Coadministration of entecavir with lamivudine, adefovir dipivoxil, or tenofovir disoproxil fumarate did not result in significant drug interactions. The effects of coadministration of BARACLUDE with other drugs that are renally eliminated or are known to affect renal function have not been evaluated, and patients should be monitored closely for adverse events when BARACLUDE is coadministered with such drugs.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term oral carcinogenicity studies of entecavir in mice and rats were carried out at exposures up to approximately 42 times (mice) and 35 times (rats) those observed in humans at the highest recommended dose of 1 mg/day.
In mouse and rat studies, entecavir was positive for carcinogenic findings. In mice, lung adenomas were increased in males and females at exposures 3 and 40 times those in humans. Lung carcinomas in both male and female mice were increased at exposures 40 times those in humans. Combined lung adenomas and carcinomas were increased in male mice at exposures 3 times and in female mice at exposures 40 times those in humans. Tumor development was preceded by pneumocyte proliferation in the lung, which was not observed in rats, dogs, or monkeys administered entecavir, supporting the conclusion that lung tumors in mice may be a species-specific event. Hepatocellular carcinomas were increased in males and combined liver adenomas and carcinomas were also increased at exposures 42 times those in humans. Vascular tumors in female mice (hemangiomas of ovaries and uterus and hemangiosarcomas of spleen) were increased at exposures 40 times those in humans. In rats, hepatocellular adenomas were increased in females at exposures 24 times those in humans; combined adenomas and carcinomas were also increased in females at exposures 24 times those in humans. Brain gliomas were induced in both males and females at exposures 35 and 24 times those in humans. Skin fibromas were induced in females at exposures 4 times those in humans.
It is not known how predictive the results of rodent carcinogenicity studies may be for humans.
Entecavir was clastogenic to human lymphocyte cultures. Entecavir was not mutagenic in the Ames bacterial reverse mutation assay using S. typhimurium and E. coli strains in the presence or absence of metabolic activation, a mammalian-cell gene mutation assay, and a transformation assay with Syrian hamster embryo cells. Entecavir was also negative in an oral micronucleus study and an oral DNA repair study in rats. In reproductive toxicology studies, in which animals were administered entecavir at up to 30 mg/kg for up to four weeks, no evidence of impaired fertility was seen in male or female rats at systemic exposures >90 times those achieved in humans at the highest recommended dose of 1 mg/day. In rodent and dog toxicology studies, seminiferous tubular degeneration was observed at exposures ≥35 times those achieved in humans. No testicular changes were evident in monkeys.
Pregnancy Category C
Reproduction studies have been performed in rats and rabbits at orally administered doses of 200 and 16 mg/kg/day and showed no embryotoxicity or maternal toxicity in rat and rabbit at doses producing systemic exposures approximately 28 and 212 times those achieved at the highest recommended dose of 1 mg/day in humans. In rats, maternal toxicity, embryo-fetal toxicity (resorptions), lower fetal body weights, tail and vertebral malformations, reduced ossification (vertebrae, sternebrae, and phalanges), and extra lumbar vertebrae and ribs were observed at exposures 3100 times those in humans. In rabbits, embryo-fetal toxicity (resorptions), reduced ossification (hyoid), and an increased incidence of 13th rib were observed at exposures 883 times those in humans. In a peri-post-natal study, no adverse effects on offspring were seen with entecavir administered orally to rats at exposures >94 times those in humans. There are no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, BARACLUDE should be used during pregnancy only if clearly needed and after careful consideration of the risks and benefits. Pregnancy Registry: To monitor fetal outcomes of pregnant women exposed to entecavir, a pregnancy registry has been established. Healthcare providers are encouraged to register patients by calling 1-800-258-4263.
Use in Racial/Ethnic Groups
Clinical studies of BARACLUDE did not include sufficient numbers of subjects from some racial/ethnic minorities (black/African American, Hispanic) to determine whether they respond differently to treatment with the drug. There are no significant racial differences in entecavir pharmacokinetics.
Mechanism of Action
Entecavir, a guanosine nucleoside analogue with activity against HBV polymerase, is efficiently phosphorylated to the active triphosphate form, which has an intracellular half-life of 15 hours. By competing with the natural substrate deoxyguanosine triphosphate, entecavir triphosphate functionally inhibits all three activities of the HBV polymerase (reverse transcriptase, rt): (1) base priming, (2) reverse transcription of the negative strand from the pregenomic messenger RNA, and (3) synthesis of the positive strand of HBV DNA. Entecavir triphosphate has an inhibition constant (Ki) for HBV DNA polymerase of 0.0012 µM. Entecavir triphosphate is a weak inhibitor of cellular DNA polymerases _, §, and _ and mitochondrial DNA polymerase _ with Ki values ranging from 18 to >160 µM.
Antiviral Activity
Entecavir inhibited HBV DNA synthesis (50% reduction, EC50) at a concentration of 0.004 µM in human HepG2 cells transfected with wild-type HBV. The median EC50 value for entecavir against lamivudine-resistant HBV (rtL180M, rtM204V) was 0.026 µM (range 0.010-0.059 µM). In contrast, no clinically relevant activity was noted against human immunodeficiency virus (HIV) type 1 (EC50 value >10 µM) grown in cell culture.
Daily or weekly entecavir treatment significantly reduced viral DNA levels (4 to 8 log10) in two relevant animal models, woodchucks chronically infected with woodchuck hepatitis virus (WHV) and ducks infected with duck HBV. Long-term studies in woodchucks demonstrated that oral weekly dosing of 0.5 mg/kg entecavir (equivalent to the 1-mg human dose) maintained viral DNA levels at undetectable levels (<200 copies/mL by PCR) for up to 3 years in 3 of 5 woodchucks. No entecavir resistance changes were detected in the HBV polymerase in any of the treated animals for up to 3 years of treatment.
The coadministration of HIV nucleoside reverse transcriptase inhibitors (NRTIs) with BARACLUDE is unlikely to reduce the antiviral efficacy of BARACLUDE against HBV or of any of these agents against HIV. In HBV combination assays in vitro, abacavir, didanosine, lamivudine, stavudine, tenofovir, or zidovudine were not antagonistic to the anti-HBV activity of entecavir over a wide range of concentrations. In HIV antiviral assays, entecavir was not antagonistic to the in vitro anti-HIV activity of these six NRTIs at >4 times the Cmax of entecavir.
Entecavir is predominantly eliminated by the kidney with urinary recovery of unchanged drug at steady state ranging from 62% to 73% of the administered dose. Renal clearance is independent of dose and ranges from 360 to 471 mL/min suggesting that entecavir undergoes both glomerular filtration and net tubular secretion.
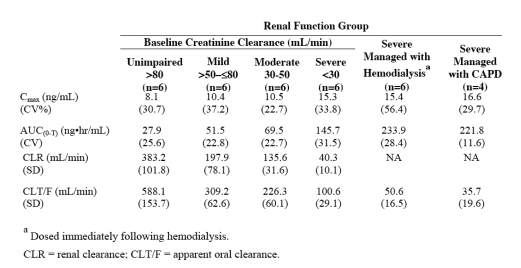
Renal impairment: The pharmacokinetics of entecavir following a single 1-mg dose were studied in patients (without chronic hepatitis B infection) with selected degrees of renal impairment, including patients whose renal impairment was managed by hemodialysis or continuous ambulatory peritoneal dialysis (CAPD). Results are shown in Table 1.
Dosage adjustment is recommended for patients with a creatinine clearance <50 mL/min, including patients on hemodialysis or CAPD.
|
|
| |
 |
| |
Hepatic impairment: The pharmacokinetics of entecavir following a single 1-mg dose were studied in patients (without chronic hepatitis B infection) with moderate or severe hepatic impairment (Child-Pugh Class B or C). The pharmacokinetics of entecavir were similar between hepatically impaired patients and healthy control subjects; therefore, no dosage adjustment of BARACLUDE is recommended for patients with hepatic impairment.
The metabolism of entecavir was evaluated in in vitro and in vivo studies. Entecavir is not a substrate, inhibitor, or inducer of the cytochrome P450 (CYP450) enzyme system. At concentrations up to approximately 10,000-fold higher than those obtained in humans, entecavir inhibited none of the major human CYP450 enzymes 1A2, 2C9, 2C19, 2D6, 3A4, 2B6, and 2E1. At concentrations up to approximately 340-fold higher than those observed in humans, entecavir did not induce the human CYP450 enzymes 1A2, 2C9, 2C19, 3A4, 3A5, and 2B6. (See CLINICAL PHARMACOLOGY: Metabolism and Elimination.) The pharmacokinetics of entecavir are unlikely to be affected by coadministration with agents that are either metabolized by, inhibit, or induce the CYP450 system. Likewise, the pharmacokinetics of known CYP substrates are unlikely to be affected by coadministration of entecavir.
The steady-state pharmacokinetics of entecavir and coadministered drug were not altered in interaction studies of entecavir with lamivudine, adefovir dipivoxil, and tenofovir disoproxil fumarate.
|
|
| |
| |
|
|
|