| |
FosAPV+LPV/r Combination Reduced LPV & APV Levels
|
| |
| |
"Combining fosamprenavir with lopinavir/ritonavir substantially reduces amprenavir and lopinavir exposure: ACTG protocol A5143 results"
AIDS: Volume 19(2) 28 January 2005
"...A5143 was designed to compare salvage regimens containing the two dual PI combinations of fosamprenavir/RTV or LPV/RTV with the combination of fosamprenavir plus LPV/RTV...
...APV and LPV exposures are significantly reduced using LPV/RTV/fosamprenavir, possibly increasing the risk of virologic failure. Consequently, A5143 was closed to enrollment... Exposure to LPV was decreased 50-60% by the addition of fosamprenavir, and exposure to amprenavir was decreased 60-70% by the addition of LPV/RTV. Ritonavir exposure in both arms did not decrease significantly... Dosing recommendations cannot be made to overcome the negative two-way interaction observed...
... Data generated in this study with fosamprenavir 700 mg and LPV/RTV 400/100 mg twice daily suggest a larger magnitude of change in APV exposure (60-70% reduction) than has been seen to date with APV 600 mg and LPV/RTV 400 mg/100 mg twice daily (0-66% reduction across studies) [7-10,13-17]. Interaction data with saquinavir combined with APV or fosamprenavir also exist. While fosamprenavir added to saquinavir/RTV resulted in a 10-24% decrease in saquinavir pharmacokinetic parameters [38], two studies with APV and saquinavir/RTV have shown either an increase of 5-30% [39] or a decrease of 74-82% [40] in saquinavir pharmacokinetic parameters. With saquinavir, changes in APV pharmacokinetic parameters ranged from an increase of 1-30% [40] to a decrease of 12-51% [39]. These findings suggest the possibility that the interactions between APV and other PI may differ from those between fosamprenavir and other PI. Additional studies with parallel designs and within-subject comparisons are needed to definitively determine this...
...Several factors may have contributed to this observed two-way interaction; concomitant medications, alterations in drug-metabolizing enzyme activity, alterations in drug transporter activity, and/or alterations in protein binding (see Author Discussion below for more discussion on this)...
...RTV is widely used to increase plasma concentrations of other PI by inhibiting their clearance. Recently, interest has emerged for using two PI in combination with RTV (i.e., triple PI combinations). Such triple PI combinations are being prescribed in clinical practice without long-term data from comparative clinical studies. Although there are theoretical advantages of better antiviral efficacy with a triple PI combination, there are potential disadvantages, including greater toxicity and complex drug-drug interactions. The safety and efficacy of triple PI combinations compared with dual PI combinations should be studied before they are incorporated into clinical practice...
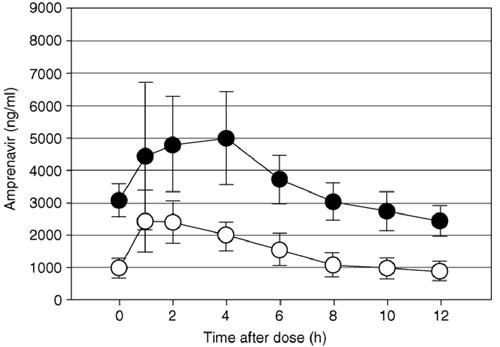
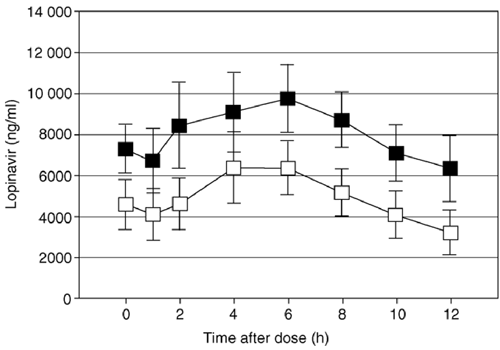
... RESULTS: Pharmacokinetic analysis. Comparison of the concentration-time plots for APV when fosamprenavir/RTV was administered alone (arm B) or in combination with LPV/RTV (arm C), revealed that APV concentrations were markedly lower in arm C (fig 1). Specifically, APV C 12 h was 69% lower (99.9% UCB, 39%; P <= 0.0001) and APV AUC0-12 h was 64% lower (99.9% UCB, 36%; P <= 0.0001) in arm C compared with arm B. LPV concentrations were also lowered by the addition of fosamprenavir to LPV/RTV compared with LPV/RTV administered alone (arm A) (Fig. 2). LPV C 12 h was 61% lower (99.9% UCB, 2%; P = 0.0001) and LPV AUC0-12 h was 48% lower (99.9% UCB, 1%; P = 0.0008) in arm C compared with arm A. There was a trend toward lower RTV concentrations with the LPV/RTV/fosamprenavir combination compared with LPV/RTV administered alone, but the P values did not cross the Peto boundary of 0.001 [C 12 h, 45% (99.9% UCB, -90%); P = 0.04; AUC0-12 h, 31% (99.9% UCB, -30%); P = 0.02]. Inference and estimates were similar when the analysis was restricted to males only, when unevaluable pharmacokinetic data were included, when comparisons restricted to prior PI exposure status were relaxed, and when non-parametric methods were used (data not shown)..."
Fig 1 Fig 2
|
|
| |
| |
 |
|
| |
| |
|
|
| |
| |
 |
|
| |
| |
Several factors may have contributed to this observed two-way interaction; concomitant medications, alterations in drug-metabolizing enzyme activity, alterations in drug transporter activity, and/or alterations in protein binding. TDF has been previously implicated in drug interactions with other antiretroviral medications, and all subjects were receiving TDF in this study. However, concentrations of APV, LPV, and RTV in arms A and B were similar to those previously observed when fosamprenavir/RTV or LPV/RTV was given without TDF [19,24]. Therefore, tenofovir is unlikely to have contributed to the observed interaction.
authors: Kashuba, Angela DMa; Tierney, Camlinb; Downey, Gerald Fb; Acosta, Edward Pc; Vergis, Emanuel Nd; Klingman, Karine; Mellors, John Wd; Eshleman, Susan Hf; Scott, Trevor Rg; Collier, Ann Ch
From the aSchool of Pharmacy, University of North Carolina, Chapel Hill, North Carolina
bSDAC/Department of Biostatistics, Harvard School of Public Health, Boston, Massachusetts
cDivision of Clinical Pharmacology, University of Alabama, Birmingham, Alabama
dDivision of Infectious Diseases, University of Pittsburgh, Pittsburgh, Pennsylvania
eHIV Research Branch, TRP, DAIDS, NIAID, NIH, Bethesda, Maryland
fDepartment of Pathology, Johns Hopkins Medical Institutions, Baltimore, Maryland
gGlaxoSmithKline, Research Triangle Park, North Carolina
hDepartment of Medicine, University of Washington School of Medicine, Seattle, Washington, USA
Abstract
Objective: To evaluate fosamprenavir/lopinavir (LPV)/ritonavir (RTV), fosamprenavir/RTV, or LPV/RTV in antiretroviral treatment-experienced patients. Lack of drug interaction data prompted a pharmacokinetic substudy to minimize subject risk.
Design: Multi-center, open-label, selectively randomized, steady-state pharmacokinetic study in HIV-infected subjects.
Methods: A planned independent interim review occurred after at least eight subjects were randomized to each arm. Subjects received twice daily LPV/RTV 400/100 mg (arm A; n = 8); fosamprenavir/RTV 700/100 mg (arm B; n = 8) or LPV/RTV/fosamprenavir 400/100/700 mg (arm C; n = 17). Plasma samples were collected over 12 h between study weeks 2 and 4. Pharmacokinetic parameters were compared based on a one-sided t-test on log-transformed data with a Peto stopping boundary (P < 0.001).
Results: Amprenavir mean area under the curve over 12 h (AUC0-12 h) and concentration at 12 h (C 12 h) (μg/ml) were, respectively, 42.7 μg × h/ml (range, 33.1-55.1) and 2.4 μg/ml (range, 1.4-3.2) in arm B and 17.4 μg × h/ml (range, 4.6-41.3) and 0.9 μg/ml (range, 0.2-2.7) in arm C: geometric mean ratio (GMR) arm C:B was 0.36 [99.9% upper confidence boundary (UCB), 0.64] and 0.31 (99.9% h UCB, 0.61), respectively (P <= 0.0001). Lopinavir AUC0-12 h and C 12 h were, respectively, 95.3 μg × h/ml (range, 60.3-119.3) and 6.3 μg/ml (range, 2.2-9.2) in arm A and 54.4 μg × h/ml (range, 23.5-112.2) and 3.0 μg/ml (range, 0.4-7.9) in arm C: GMR arm C:A of 0.52 (99.9% UCB, 0.89) and 0.39 (99.9% UCB, 0.98), respectively (P <= 0.0008). Ritonavir exposure was not significantly different between arms.
Conclusion: APV and LPV exposures are significantly reduced using LPV/RTV/fosamprenavir, possibly increasing the risk of virologic failure. Consequently, A5143 was closed to enrollment.
Author Discussion
RTV is widely used to increase plasma concentrations of other PI by inhibiting their clearance. Recently, interest has emerged for using two PI in combination with RTV (i.e., triple PI combinations). Such triple PI combinations are being prescribed in clinical practice without long-term data from comparative clinical studies. Although there are theoretical advantages of better antiviral efficacy with a triple PI combination, there are potential disadvantages, including greater toxicity and complex drug-drug interactions. The safety and efficacy of triple PI combinations compared with dual PI combinations should be studied before they are incorporated into clinical practice.
A5143 was designed to compare salvage regimens containing the two dual PI combinations of fosamprenavir/RTV or LPV/RTV with the combination of fosamprenavir plus LPV/RTV. At the time A5143 was designed, data were not available on the optimal dosing of LPV/RTV combined with fosamprenavir in HIV-infected subjects. To guard patient safety, a pharmacokinetic substudy of A5143 with real-time pharmacokinetic analyses was designed and an interim analysis planned. Enrollment into A5143 was initially restricted to subjects agreeing to participate in the pharmacokinetic substudy. This pharmacokinetic study demonstrated a significant two-way drug interaction between LPV/RTV and fosamprenavir. Exposure to LPV was decreased 50-60% by the addition of fosamprenavir, and exposure to amprenavir was decreased 60-70% by the addition of LPV/RTV. Ritonavir exposure in both arms did not decrease significantly.
Several factors may have contributed to this observed two-way interaction; concomitant medications, alterations in drug-metabolizing enzyme activity, alterations in drug transporter activity, and/or alterations in protein binding. TDF has been previously implicated in drug interactions with other antiretroviral medications, and all subjects were receiving TDF in this study. However, concentrations of APV, LPV, and RTV in arms A and B were similar to those previously observed when fosamprenavir/RTV or LPV/RTV was given without TDF [19,24]. Therefore, tenofovir is unlikely to have contributed to the observed interaction.
Induction of the activity of the cytochrome P450 3A subfamily of enzymes (CYP3A) leading to decreased concentrations of CYP3A substrates has been described for APV, both in vitro [25] and in healthy volunteers [26,27]. Because the primary LPV/RTV metabolic pathway is through the CYP3A subfamily, induction of CYP3A enzyme activity by APV may account for decreased LPV concentrations. This is supported by recent data generated in two studies of triple PI use in healthy volunteers, demonstrating that decreased LPV concentrations could be overcome by adding either an extra 100 mg RTV twice daily or an extra LPV/RTV capsule (533/133 mg twice daily) to the fosamprenavir/LPV/RTV regimen [21]. APV is also predominantly metabolized by CYP3A, and LPV/RTV has been implicated in significantly decreasing CYP3A substrate concentrations [24,28]. Kumar et al. [29] demonstrated that combining LPV with RTV in vitro resulted in a 10-fold decrease in the inhibitory potency of RTV (with an average inhibition constant of approximately 800 ng/ml). However, unlike the situation with LPV, two healthy volunteer studies showed that the fosamprenavir interaction was not overcome by extra doses of RTV or by doubling the dose of fosamprenavir [21]. Therefore, it is unlikely that a CYP3A-mediated interaction is solely responsible for this study's findings on APV concentrations.
Another potential contributor to lowered drug exposure is the drug transporter P-glycoprotein. This efflux transporter can decrease drug concentrations by pumping drug out of the enterocyte during absorption, or pumping drug out of the hepatocyte into the biliary cannaliculi during circulation. APV and LPV are both substrates for P-glycoprotein, and recent investigations suggest a potential association between P-glycoprotein genotype and the pharmacokinetics of certain PI drugs [30]. However, a complete understanding is still evolving. In vitro studies do not support APV increasing P-glycoprotein activity [31,32], and it is controversial whether or not LPV/RTV appreciably induces P-glycoprotein [31,33,34]. Therefore, it is unlikely that P-glycoprotein solely mediated this interaction.
Protein binding displacement may result in altered pharmacokinetics of drugs; APV and LPV bind to α1-acid glycoprotein with high affinity [24,35]. One recent clinical investigation compared total and unbound APV concentrations in nine patients first receiving APV 600 mg twice daily plus 100-200 mg RTV twice daily, and then adding LPV/RTV 400/100 mg twice daily [9]. These investigators determined the median unbound fraction of APV to be significantly higher after the addition of LPV/RTV (11.4% with LPV/RTV versus 8.9%; P = 0.03). However, the median unbound APV AUC0-10 h significantly declined after the addition of LPV/RTV (1434 ng × h/ml with LPV/RTV versus 2436 ng × h/ml; P = 0.004). Therefore, displacement from protein binding alone also does not account for this interaction [36].
Comparison of the pharmacokinetic parameters of fosamprenavir alone and in combination with LPV/RTV suggested that the interaction could be mediated during absorption, since a dramatic effect was noted on maximum plasma concentrations. To investigate this further, a healthy volunteer study was conducted in parallel to A5143 and evaluated physical separation of fosamprenavir and LPV/RTV dosing by 4-12 h [37]. Separating the administration of fosamprenavir and LPV/RTV doses by up to 12 h (administering fosamprenavir/RTV 1400 mg/200 mg once daily in the morning and LPV/RTV 800 mg/200 mg once daily in the evening) did not significantly increase APV concentrations, compared with taking fosamprenavir concurrently with LPV/RTV (although LPV concentrations were increased).
Data generated in this study with fosamprenavir 700 mg and LPV/RTV 400/100 mg twice daily suggest a larger magnitude of change in APV exposure (60-70% reduction) than has been seen to date with APV 600 mg and LPV/RTV 400 mg/100 mg twice daily (0-66% reduction across studies) [7-10,13-17]. Interaction data with saquinavir combined with APV or fosamprenavir also exist. While fosamprenavir added to saquinavir/RTV resulted in a 10-24% decrease in saquinavir pharmacokinetic parameters [38], two studies with APV and saquinavir/RTV have shown either an increase of 5-30% [39] or a decrease of 74-82% [40] in saquinavir pharmacokinetic parameters. With saquinavir, changes in APV pharmacokinetic parameters ranged from an increase of 1-30% [40] to a decrease of 12-51% [39]. These findings suggest the possibility that the interactions between APV and other PI may differ from those between fosamprenavir and other PI. Additional studies with parallel designs and within-subject comparisons are needed to definitively determine this.
In the current study, C 12 h of APV in the fosamprenavir/LPV/RTV arm ranged from 224 to 2683 ng/ml. Many patients achieved APV concentrations greater than those achieved when APV was administered as 1200 mg twice daily without RTV [35]. However, these concentrations may be lower than desired for patients who have failed one or two previous PI regimens and who have virus with multiresistant to PI drugs. Analysis of virological responses to the treatment arms in A5143 will be performed once all subjects have completed their follow-up visits. However, because enrollment in the trial was stopped prematurely, this trial has power to detect only large differences.
This study demonstrated an important two-way pharmacokinetic interaction between LPV/RTV and fosamprenavir. The study design allowed identification of this drug-drug interaction in real-time and provided information that will likely influence the choice of PI regimens in treatment-experienced patients on a failing regimen. Dosing recommendations cannot be made to overcome the negative two-way interaction observed. Further investigation of this interaction is needed to identify the mechanisms involved and to develop dosing strategies that may allow this combination to be used effectively.
Introduction
Developing effective therapies for treatment-experienced patients with persistent viremia on a failing regimen is an important objective for the management of patients with HIV-1 infection. One strategy attempts to improve the activity of regimens by increasing antiretroviral drug concentrations. The use of a pharmacokinetic-enhancing agent such as ritonavir (RTV), which increases the drug concentrations of other HIV-1 protease inhibitors (PI) by interfering with their metabolism, has been particularly successful. Recently, triple PI regimens, which include two active PI combined with RTV as a pharmacokinetic enhancing agent, have been examined for activity in treatment-experienced patients. One such combination in use is lopinavir (LPV)/RTV (Kaletra; Abbott Laboratories, Abbott Park, Illinois, USA) and amprenavir (APV) (Agenerase; GlaxoSmithKline, Research Triangle Park, North Carolina, USA). The pharmacokinetics of the LPV/RTV 400/100 mg plus APV 600 mg combination taken twice daily have been reported by a number of investigators [1-17]. Many of these were small, retrospective evaluations, which often compared different dosing regimens. In general, APV appears to lower LPV concentrations, and in some patients LPV may lower APV concentrations. Point estimates for the magnitude of the interaction vary from a 0% decline to 66% decline for both APV and LPV.
Fosamprenavir (Lexiva; GlaxoSmithKline) is the calcium phosphate ester prodrug of APV and has been developed to deliver APV with smaller and fewer tablets. Fosamprenavir is converted to APV in the brush-border of the gastrointestinal tract, with <0.6% exposure of fosamprenavir detectable in the plasma [18]. A fosamprenavir/ RTV 700 mg/100 mg twice daily regimen results in APV drug exposures similar to or greater than APV/RTV 600 mg/100 mg twice daily [19,20].
The Adult AIDS Clinical Trials Group (ACTG) Study A5143 was a randomized comparison of LPV/RTV or fosamprenavir/RTV versus LPV/RTV plus fosamprenavir, each combined with tenofovir disoproxil fumarate (TDF) and one or two nucleoside reverse transcriptase inhibitors, in HIV-1-infected subjects with prior treatment experience and virologic failure on their current regimen. When A5143 was designed, data were not available on the pharmacokinetics of the combination of fosamprenavir 700 mg twice daily with LPV/RTV 400 mg/100 mg twice daily in HIV-infected subjects. This prompted the design of A5147s, a pharmacokinetic substudy of A5143, with a primary objective of determining the effects of fosamprenavir coadministration on the pharmacokinetics of LPV/RTV and to determine the effects of LPV/RTV on the pharmacokinetics of amprenavir in the first 20 subjects randomized to each study arm. Prior to finalization of the study, data became available from a fosamprenavir-LPV/RTV pharmacokinetic interaction study in healthy seronegative human volunteers [21]. However, these data did not include the dosing strategy planned for this protocol. Since the magnitude of the potential drug interaction in HIV-infected subjects receiving standard doses of fosamprenavir and LPV/RTV was still unknown, study development proceeded. As a consequence of the new data, an interim analysis was added to the protocol to occur after eight evaluable subjects were enrolled in each study arm. This report describes the results from the interim analysis that led to closure of A5143 to further enrollment.
Methods
Subjects
Male and female HIV-infected subjects at least 18 years of age were considered for enrollment. Inclusion criteria were plasma HIV RNA >5000 copies/ml after at least 12 weeks on their current antiretroviral regimen, at least 12 weeks of prior PI exposure, a minimum total duration of antiretroviral therapy of 1 year, and, for the first 36 subjects enrolled, plasma HIV-1 at screening susceptible to APV (<=2-fold higher than the reference strain) and LPV (<=2.5 higher) as determined by Virtual Phenotype (VIRCO, Mechelen, Belgium). Exclusion criteria included subjects with >7 days of prior exposure to both APV (or fosamprenavir) and LPV, significant medical conditions or laboratory parameter abnormalities, current history of alcohol or drug abuse, current use of any cytochrome P450 inhibitors or inducers, and a history of allergy to any study medication. Sexually active females were required to have a negative pregnancy test and to use barrier contraception during the study. All subjects provided written informed consent before any study procedures.
Materials
LPV/RTV was administered orally at the approved dose of 400/100 mg twice daily. Fosamprenavir was administered orally at the approved dose of 700 mg twice daily together with 100 mg of RTV. TDF was administered orally at the dose of 300 mg once daily. One or two other NRTI drugs were chosen prior to randomization by the subject in conjunction with his or her primary care provider and the site investigator. Subjects were instructed to take LPV, RTV, and TDF with food. Subjects took fosamprenavir and RTV simultaneously during coadministration, and took LPV/RTV and fosamprenavir simultaneously during coadministration. The timing of the last four doses of antiretroviral drug prior to the pharmacokinetic assessment was collected through subject diaries. During inpatient visits, a standardized breakfast (500-700 kcal; 33-35% fat) was served 30 min prior to pharmacokinetic sampling and was to be completed within 30 min. No grapefruit juice or grapefruit products were allowed. Blood samples for pharmacokinetic analysis were collected in 10 ml ethylenediaminetetraacetic acid tubes.
Study design
The study was a selectively randomized, open-label, parallel-arm, multicenter study. Following a screening visit and confirmation that all entry criteria were met, subjects who were naive to both APV and LPV/RTV were randomized 1:1:2 to one of the three study arms. Subjects with experience to one of the two PI were randomized 1:1 to either the RTV-enhanced single PI to which they were naive (arm A or B) or to the LPV/RTV/fosamprenavir arm (arm C).
Blood samples for evaluation of steady-state concentrations of APV, LPV, and RTV were obtained during the week 2 study visit (14-28 days after starting study treatment) following an observed, simultaneous administration of the first morning doses of the PI, TDF, and the NRTI(s). The time and date of the previous four doses of PI were obtained and recorded. Plasma samples were obtained before dose and at 1, 2, 4, 6, 8, 10, and 12 h after administration. Lunch was permitted after the 4 h sample.
The toxicity grading scale of the AIDS Clinical Trials Group was used for the reporting of clinical and laboratory adverse events [22].
Analytical methods
Following blood collection, samples were kept on ice until processed (within 90 min of collection). Plasma was separated by centrifugation at 800 × g for 10 min, transferred to a polypropylene cryovial, and frozen at -20 °C to -70 °C until shipment. Samples were shipped within 2 working days of collection to the Antiviral Pharmacology Laboratory at the University of Alabama at Birmingham for analysis. Samples were analyzed within 14 days of collection, and the pharmacokinetic parameters were calculated within 48 h and transferred to the central database within 72 h.
APV, LPV, and RTV plasma concentrations were assayed using a validated liquid chromatography method using a Waters Alliance 2695 Separations Module with a Waters 2487 ultraviolet detector set to 210 nm [23]. Briefly, each 0.2 ml plasma sample was mixed, vortexed, and centrifuged with A-86093 internal standard (obtained from Abbott laboratories) and 1 ml tert-butyl methyl ether. Samples were frozen for 15 min and the organic layer removed to a clean vial and evaporated to dryness under nitrogen and low heat. Dried residues were reconstituted in 100 μl mobile phase. Samples were run on a Supelguard Discovery C8 analytical column. The mobile phase conditions consisted of (A) 25 mmol/l potassium phosphate pH 3.1, (B) acetonitrile, and (C) methanol. Separation was facilitated via gradient elution at 1.5 ml /min flow rate with initial mobile phase concentrations of 55% A, 25% B and 20% C, and final concentrations of 55% A, 45% B, and 0% C. Calibration curves for APV, LPV, and RTV ranged from 50.0 to 10 000 ng/ml. Inter- and intraday variance was <15% and <10% respectively.
Data analysis and statistical methods
Pharmacokinetic parameters were derived from plasma concentrations using non-compartmental methods (Model 200, WinNonlin Pro 4.0.1; Pharsight Corporation, Cary, North Carolina, USA). To be evaluable, data must have been available from the samples taken predose, at 6 and 12 h after dosing, and for at least three of the five other samples. The log-linear trapezoidal method was used to calculate the area under the concentration-time curve from 0 to 12 h (AUC0-12 h), wherein the linear trapezoidal rule was used up to the maximum concentration and the logarithmic trapezoidal rule was used after this.
Statistical comparisons were performed on the (natural) logarithmic scale. To preserve the effect of randomization, comparisons between treatment arms were restricted to subjects who were eligible to be randomized to both of the arms being compared. For example, comparisons of APV concentrations between arms B and C were restricted to subjects randomized to arm C who were APV naive at entry and thus were comparable with subjects randomized to arm B. Mean AUC0-12 h or concentration at 12 h (C 12 h) were compared with a one-sided t-test, assuming unequal variances. To ensure that the conclusion was robust to the deviation from normality assumption, the non-parametric Wilcoxon Rank Sum test (exact) was also used as a confirmatory test. Geometric mean ratios (GMR) and upper 99.9% confidence intervals (99.9% UCB) were also calculated for AUC0-12 h and C 12 h.
A priori sample size estimates indicated that 20 evaluable subjects per arm would be expected to provide 80% power to detect a 23-30% reduction in the mean steady-state C 12 h in the combination arm (arm C) compared with the fosamprenavir/RTV-only arm (arm B) and the LPV/RTV-only arm (arm A), respectively. This assumed a one-sided t-test with a 0.05 significance level and means in the fosamprenavir/RTV and LPV/RTV arms of 2.22 μg/ml (SD, 1.39) and 5.68 μg/ml (SD, 1.57), respectively (obtained from normal volunteer studies performed by GlaxoSmithKline). However, because the magnitude of potential interactions was unknown, a planned interim analysis was performed after at least eight subjects from each treatment arm had evaluable pharmacokinetic data. An independent interim review committee appointed by the AACTG performed the review of these data. Stopping guidelines specified a Peto stopping boundary (P < 0.001) for comparison between treatment arms. All statistical analyses were carried out using SAS statistical package version 8.2 (SAS Institute, Cary, North Carolina, USA) and R version 1.6.2 ( http://www.r-project.org ).
Results
Baseline characteristics
Forty-four subjects were enrolled in A5143 and the substudy A5147s between October 2002 and June 2003. When the study database was frozen for the interim analysis, pharmacokinetic data for eight subjects were not available, and three subjects (one in each arm) had unusable pharmacokinetic data (one subject had no detectable drug concentrations; one subject did not have a pharmacokinetic blood sample 12 h after the observed dose, and one had a pre-dose pharmacokinetic blood sample obtained more than 1 h prior to the dose time). Therefore, pharmacokinetic data from a total of 33 subjects were used in the interim analysis, consisting of eight subjects in each of arms A and B and 17 subjects in arm C. The majority of enrolled subjects were male (88%) and white (52%). Median age was 42 years (25-75th percentile, 37-47), median weight 76.8 kg (64.5-84.5), and median height 1.7 m (1.60-1.8) (Table 1). All 33 subjects had plasma-derived HIV-1 from the screening visit that was susceptible to APV and LPV as determined by Virtual Phenotype™.
PK Results shown at beginning of this report.
Adverse events
From initiation of therapy to the time of the pharmacokinetic analysis, study medications were generally well tolerated. Three subjects experienced grade 1 or higher transient, self-limited symptoms: one subject on arm C had grade 2 diarrhea; another subject on arm C had grade 3 fever; and one subject on arm A experienced grade 3 rash, fever, edema, and discomfort. However, no laboratory abnormalities were noted.
References
1 Baldini F, Rizzo MG, Hoetelmans R, Murri R, Giambenedetto SD, Cingolani A, et al. prospective study of deep salvage therapy with lopinavir/r, amprenavir, and NRTIs: final 24-week data, pharmacokinetics, and association of drug levels/drug susceptibility with virologic response. Ninth Conference on Retroviruses and Opportunistic Infections. Seattle, February 2002 [abstract 423-W].
2 Bertz R, Foit C, Burt D, Hsu A, Chiu Y-L, Chira T, et al. Assessment of the multiple dose pharmacokinetic interaction between Kaletra (lopinavir/ritonavir) and amprenavir in healthy volunteers. Third International Workshop on Clinical Pharmacology of HIV Therapy. Washington, DC, April 2002 [poster 7.6].
3 Bertz R, Foit C, Ashbrenner E, Ashbrenner D, Burt LA, Williams T, Chira et al. Effect of amprenavir on the steady-state pharmacokinetic of lopinavir/ritonavir in HIV+ and healthy subjects. 42nd Interscience Conference on Antimicrobial Agents and Chemotherapy. San Diego, 2002 [poster A-1823].
4 Nadler J, Laartz B, Le T. Immunologic and virologic benefit accruing to patients treated with amprenavir (APV) + Kaletra™ (lopinavir [LPV]/ritonavir [RTV]. 40th Infectious Diseases Society of America. Chicago, October 2002 [poster 446].
5 Reynolds H, Gibbons S, Tija J. The pharmacokinetic interaction of lopinavir/ritonavir and amprenavir in clinical practice. XIV International Conference on AIDS. Barcelona, July 2002 [poster TuPeB4560].
6 Vanig T, Garg V, Brill M. Decreased plasma concentrations of amprenavir and lopinavir in HIV patients on this combination. XIV International Conference on AIDS. Barcelona, July 2002 [poster MoPpB2009].
7 Wynn Vezina H, Brundage R, Bushman L, Fletcher C. Pharmacologic management of the drug-drug interaction between lopinavir/ritonavir and amprenavir. Eleventh Conference on Retroviruses and Opportunistic Infections. San Francisco, February 2004 [poster 609].
8 DeJesus E, Oritz R, Bellow N, Grossman H, Anderson R, Brothers C, et al. An investigation to determine amrpenavir and lopinavir/ritonavir plasma trough concentrations when co-administered. Third International Workshop on Clinical Pharmacology of HIV Therapy. Washington, DC, April 2002 [poster 7.1].
9 Taburet AM, Raguin G, Le Tiec C, Droz C, Barrail A, Vincent I, et al. Interactions between amprenavir and the lopinavir-ritonavir combination in heavily pretreated patients infected with human immunodeficiency virus. Clin Pharmacol Ther 2004; 75:310-323.
10 Gatti G, de Pascalis C, de Luca A, Di Biagio A, Alessandrini A, Bartolacci V, et al. Pharmacokinetics and virologic outcome with amprenavir, amprenavir/ritonavir or amprenavir/lopinavir/ritonavir. Third International Workshop on Clinical Pharmacology in HIV Therapy. Washington, April 2002 [poster 3.8].
11 Basso S, Solas C, Quinson AM, Ravaux I, Poizot-Martin I, Bacconier J, et al. Pharmacokinetic interaction between lopinavir/r and amprenavir in salvage therapy. J Acquir Immune Defic Syndr 2002; 31:115-117.
12 Mauss S, Schmutz G, Kuschak D. Unfavourable interaction of amprenavir and lopinavir in combination with ritonavir? AIDS 2002; 16:296-297.
13 Mauss S, Scholten S, Wolf E, Berger F, Schmutz G, Jaeger H, et al. A prospective, controlled study assessing the effect of lopinavir on amprenavir concentrations boosted by ritonavir. HIV Med 2004; 5:15-17.
14 de Luca A, Baldini F, Cingolani A, Di Giambenedetto S, Hoetelmans RM, Cauda R. Deep salvage with amprenavir and lopinavir/ritonavir: correlation of pharmacokinetics and drug resistance with pharmacodynamics. J Acquir Immune Defic Syndr 2004; 35:359-366.
15 Peytavin G, Lamotte C, Duval X, Matheron S, Boue F, de Truchis P, et al. Amprenavir (APV) concentrations are dramatically decreased by association with ABT378/r in HIV-infected patients. Second International Workshop on Clinical Pharmacology of HIV Therapy. Noordwik, April 2001 [poster 1.14].
16 Meynard J, Poirier J, Guiard-Schmid J, Morand-Joubert L, Jacquemet N, Rozenbaum W, et al. Impact of ABT 378/r on the amprenavir plasma concentrations in HIV-experienced patients treated by the association of APV-ABT 378/r. 41st Interscience Conference on Antimicrobial Agents and Chemotherapy. Chicago, September 2001 [a bstract I-1736].
17 Zilly M, Langmann P, Vaath T, Winzer R, Heinz W, Klinker H. Effects of efavirenz and/or lopinavir/r on amprenavir plasma levels in HAART. Third International Workshop on Clinical Pharmacology of HIV Therapy. Washington DC, April 2002 [abstract 7.8].
18 Furfine ES, Baker CT, Hale MR, Reynolds DJ, Salisbury JA, Searle AD, et al. Preclinical pharmacology and pharmacokinetics of GW433908, a water-soluble prodrug of the human immunodeficiency virus protease inhibitor amprenavir. Antimicrob Agents Chemother 2004; 48:791-798.
19 GlaxoSmithKline Lexiva Product Information. Research Triangle Park, NC: GlaxoSmithKline; 2003.
20 Goujard C, Vincent I, Meynard JL, Choudet N, Bollens D, Rousseau C, et al. Steady-state pharmacokinetics of amprenavir coadministered with ritonavir in human immunodeficiency virus type 1-infected patients. Antimicrob Agents Chemother 2003; 47:118-123.
21 Wire M, Naderer O, Masterman A, Lou Y, Stein D. The pharmacokinetic interaction between GW433908 and lopinavir/ritonavir (APV10011 and APV10012). Eleventh Conference on Retroviruses and Opportunistic Infections. San Francisco, February 2004 [poster 612].
22 AIDS Clinical Trials Group. Table of Grading Severity of Adult Adverse Experiences. Rockville MD: Division of AIDS, National Institute of Allergy and Infectious Diseases; 1996.
23 Turner ML, Reed-Walker K, King JR, Acosta EP. Simultaneous determination of nine antiretroviral compounds in human plasma using liquid chromatography. J Chromatogr B: Analyt Technol Biomed Life Sci 2003; 784:331-341.
24 Abbott Laboratories. Kaletra Product Information. Abbott Park, IL: Abbott Laboratories; 2003.
25 Huang L, Wring SA, Woolley JL, Brouwer KR, Serabjit-Singh C, Polli JW. Induction of P-glycoprotein and cytochrome P450 3A by HIV protease inhibitors. Drug Metab Dispos 2001; 29:754-760.
26 Justesen U, Klitgaard N, Brosen K. Amprenavir is an effective inducer of delavirdine metabolism: a steady-state pharmacokinetic interaction study between amprenavir and delavirdine in healthy volunteers. Ninth Conference on Retroviruses and Opportunistic Infections. Seattle, February 2002. [poster 442W].
27 Tran JQ, Petersen C, Garrett M, Hee B, Kerr BM. Pharmacokinetic interaction between amprenavir and delavirdine: evidence of induced clearance by amprenavir. Clin Pharmacol Ther 2002; 72:615-626.
28 Hsu A, Isaacson J, Brun S, Bernstein B, Lam W, Bertz R, et al. Pharmacokinetic-pharmacodynamic analysis of lopinavir-ritonavir in combination with efavirenz and two nucleoside reverse transcriptase inhibitors in extensively pretreated human immunodeficiency virus-infected patients. Antimicrob Agents Chemother 2003; 47:350-359.
29 Kumar GN, Dykstra J, Roberts EM, Jayanti VK, Hickman D, Uchic J, et al. Potent inhibition of the cytochrome P-450 3A-mediated human liver microsomal metabolism of a novel HIV protease inhibitor by ritonavir: a positive drug-drug interaction. Drug Metab Dispos 1999; 27:902-908.
30 Fellay J, Marzolini C, Meaden ER, Back DJ, Buclin T, Chave JP, et al. Response to antiretroviral treatment in HIV-1-infected individuals with allelic variants of the multidrug resistance transporter 1: a pharmacogenetics study. Lancet 2002; 359:30-36.
31 Chandler B, Almond L, Ford J, Owen A, Hoggard P, Khoo S, et al. The effects of protease inhibitors and nonnucleoside reverse transcriptase inhibitors on P-glycoprotein expression in peripheral blood mononuclear cells in vitro. J Acquir Immune Defic Syndr 2003; 33:551-556.
32 Kim RB. Drug transporters in HIV therapy. Top HIV Med 2003; 11:136-139.
33 Vishnuvardhan D, Moltke LL, Richert C, Greenblatt DJ. Lopinavir: acute exposure inhibits P-glycoprotein; extended exposure induces P-glycoprotein. AIDS 2003; 17:1092-1094.
34 Ford J, Meaden ER, Hoggard PG, Dalton M, Newton P, Williams I, et al. Effect of protease inhibitor-containing regimens on lymphocyte multidrug resistance transporter expression. J Antimicrob Chemother 2003; 52:354-358.
35 GlaxoSmithKline. Agenerase Product Information. Research Triangle Park, NC: GlaxoSmithKline; 2004.
36 MacKichan J. Influence of protein binding and use of unbound (free) drug concentrations. In: Applied Pharmacokinetics, Principles of Therapeutic Drug Monitoring. Edited by WE Evans, J Schentag, WJ Jusko. Vancouver, WA: Applied Therapeutics; 1992:5-1-5-41.
37 Corbett A, Davidson L, Park J, Patterson K, Eron J, Ngo L, et al. Dose Separation strategies to overcome the pharmacokinetic interaction of a triple protease inhibitor regimen containing fosamprenavir lopinavir, and ritonavir. Eleventh Conference on Retroviruses and Opportunistic Infections. San Francisco, February 2004.
38 Boffito M, Dickinson L, Hill A, Nelson M, Moyle G, Higgs C, et al. Steady state pharmacokinetics of saquinavir hard gel/fosamprenavir 1000/700 plus 100 mg and 200 mg of ritonavir twice daily in HIV+ patients. Eleventh Conference on Retroviruses and Opportunistic Infections. San Francisco, February 2004. [poster 608].
39 Wolfe P, Anderson P, Gunawan S. Co-administration of amprenavir does not lower plasma levels of ritonavir-boosted saquinavir. Third International Workshop on Clinical Pharmacology of HIV Therapy. Washington, April 2002 [poster 7.11].
40 Corbett AH, Eron JJ, Fiscus SA, Rezk NL, Kashuba AD. The pharmacokinetics, safety, and initial virologic response of a triple-protease inhibitor salvage regimen containing amprenavir, saquinavir, and ritonavir. J Acquir Immune Defic Syndr 2004; 36:921-928.
|
|
| |
| |
|
|
|