| |
CCR5 deficiency increases risk of symptomatic West Nile virus infection
|
| |
| |
Journal of Experimental Medicine
JAN 2006
".....Our data indicate that immunocompromised patients could be particularly susceptible to symptomatic WNV infection if CCR5 function is missing or blocked...."
".....Our results have important implications regarding the potential safety of CCR5-blocking agents now under development for the treatment of HIV/AIDS. Clinical care of individuals taking these medicines while residing in WNV-endemic areas may mandate strict measures to limit mosquito exposure and a high index of suspicion for symptoms consistent with WNV...."
William G. Glass,1 David H. McDermott,1 Jean K. Lim,1
Sudkamon Lekhong,2 Shuk Fong Yu,2 William A. Frank,3
John Pape,4 Ronald C. Cheshier,2 and Philip M. Murphy1
1Laboratory of Molecular Immunology, National Institute of Allergy and Infectious Diseases, National Institutes of Health,
Bethesda, MD 20892
2Bureau of State Laboratory Services and 3Bureau of Epidemiology and Disease Control Services,
Arizona Department of Health Services, Phoenix, AZ 85007
4Colorado Department of Public Health and Environment, Denver, CO 80246
ABSTRACT
West Nile virus (WNV) is a reemerging pathogen that causes fatal encephalitis in several species, including mouse and human. Recently, we showed that the chemokine receptor CCR5 is critical for survival of mice infected with WNV, acting at the level of leukocyte trafficking to the brain. To test whether this receptor is also protective in man, we determined
the frequency of CCR5delta32, a defective CCR5 allele found predominantly in Caucasians, in two independent cohorts of patients, one from Arizona and the other from Colorado, who had laboratory-confi rmed, symptomatic WNV infection. The distribution of CCR5delta32 in a control population of healthy United States Caucasian random blood donors was in Hardy-Weinberg equilibrium and CCR5delta32 homozygotes represented 1.0% of the total group (n = 1,318). In contrast, CCR5delta32 homozygotes represented 4.2% of Caucasians in the Arizona cohort (odds ratios [OR] = 4.4 [95% confi dence interval [CI], 1.6-11.8], P = 0.0013) and 8.3% of Caucasians in the Colorado cohort (OR = 9.1 [95% CI,3.4-24.8], P < 0.0001). CCR5delta32 homozygosity was signifi cantly associated with fataloutcome in the Arizona cohort (OR = 13.2 [95% CI, 1.9-89.9], P = 0.03). We conclude
that CCR5 mediates resistance to symptomatic WNV infection. Because CCR5 is also the major HIV coreceptor, these findings have important implications for the safety of CCR5- blocking agents under development for HIV/AIDS.
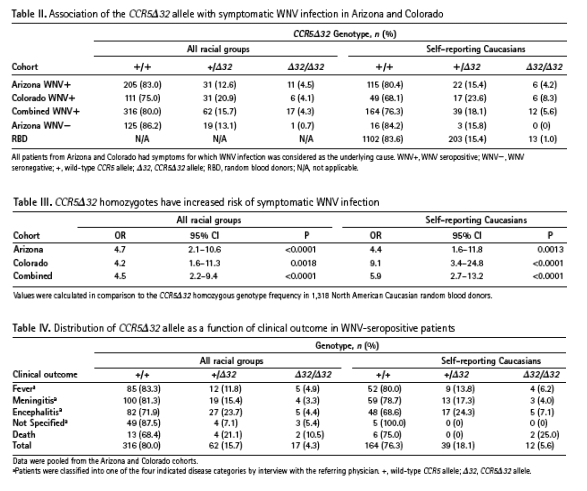
The present study provides the fi rst evidence that the defective CCR5 allele CCR5_32 is a risk factor for symptomatic WNV infection, the fi rst genetic risk factor identifi ed for this disease. The association was restricted to CCR5_32
homozygotes and was very strong, similar in magnitude to the association we and others have previously reported for CCR5_32 homozygosity with the exposed, uninfected HIV resistance phenotype (e.g., OR = 6.04 [95% CI, 1.42-25.7], P = 0.02, in reference 15). Although our results are based on retrospective analysis, they are unlikely to be because of chance for six reasons. First, the results are statistically strong (an OR greater than four). Second, the results were obtained in two independent as well as geographically and temporally distinct cohorts. Third, CCR5_32 is a complete lossof- function mutation and therefore homozygous individuals completely lack functional CCR5. Fourth, WNV infection
is associated with infi ltration of T cells and macrophages (cell types known to express CCR5) into brains of patients infected with WNV, suggesting biologically plausible involvement of CCR5 in regulating leukocyte migration to the
brains of infected patients (19, 20). Fifth, WNV infection of mice induces expression of the CCR5 ligand CCL5 and accumulation of CCR5+ leukocytes in the brain (12, 13). Sixth, WNV infection in CCR5_/_ mice is uniformly fatal (12).
Together these results imply that wild-type CCR5 functions as a host defense factor in WNV infection in man. CCR5 could potentially restrict WNV infection at the level of initial infection. This is not addressed by our retrospective study design and may not be feasible to resolve prospectively by measuring the association of CCR5_32 homozygosity with asymptomatic WNV infection because this genotype and WNV infection are both uncommon in the general population. More likely, CCR5 restricts disease progression after initial infection so that its absence results in an increased likelihood of an infected patient coming to medical attention as a symptomatic case. Our data are consistent with this interpretation, and even suggest that a fatal outcome is more likely in the absence of CCR5 (Fig. 1). Additional work will be needed to more critically address the latter point because the positive association we observed between CCR5 deficiency and death in the Arizona cohort, although statistically significant, was based on only two cases, and could not be detected in the Colorado cohort, possibly because of its smaller size.
Another limitation of our study was the incomplete information about the racial background of cases (Table I), which is needed to more precisely quantitate risk because CCR5_32 is primarily found in Caucasians. We have addressed this by
consistently applying a conservative analysis of the data that has enabled us to test boundary conditions for quantitating the strength of the genotype-disease association. Larger prospective studies will be needed to more precisely quantitate risk as well as to further defi ne mechanisms in man.
Our data indicate that immunocompromised patients could be particularly susceptible to symptomatic WNV infection if CCR5 function is missing or blocked. Additional studies will be needed to address this issue, which should include close monitoring of AIDS patients treated with antagonists now in advanced stages of development targeting the HIV coreceptor activity of CCR5. Such agents are intended to imitate the homozygous CCR5_32 genetic defect and could render AIDS patients particularly vulnerable to severe and possibly fatal WNV infection.
In summary, we have established that homozygous CCR5_32 is a strong host genetic risk factor for symptomatic laboratory-confirmed WNV infection, the first one identified for this disease. In contrast, the homozygous CCR5_32 genotype has previously been strongly associated with resistance to HIV. These genetic data imply that CCR5 plays opposite roles in HIV and WNV infection, facilitating the former and restricting the latter. Our results have important implications regarding the potential safety of CCR5-blocking agents now under development for the treatment of HIV/AIDS. Clinical care of individuals taking these medicines while residing in WNV-endemic areas may mandate strict measures to limit mosquito exposure and a high index of suspicion for symptoms consistent with WNV.
|
|
| |
| |
|
|
|