| |
Rapid Emergence of Enfuvirtide Resistance in HIV-1-Infected Patients: Results of a Clonal Analysis
|
| |
| |
JAIDS Journal of Acquired Immune Deficiency Syndromes: Volume 43(1) September 2006 pp 60-64
[Clinical Science: Brief Report]
Lu, Jing MD*; Deeks, Steven G. MD; Hoh, Rebecca RD; Beatty, George MD; Kuritzkes, Benjamin A. MPH*; Martin, Jeffrey N. MPH; Kuritzkes, Daniel R. MD*
From the *Section of Retroviral Therapeutics, Brigham and Women's Hospital; and Division of AIDS, Harvard Medical School, Boston, MA; Department of Medicine, University of California-San Francisco; and San Francisco General Hospital, San Francisco, CA; and Department of Epidemiology and Biostatistics, University of California-San Francisco, San Francisco, CA.
The study authors report: T20 (Fuzeon resistance can emerge quickly like 3Tc or a NNRTI, so it is crucial to use enough drugs in T20 containing regimen to which the patient is sensitive in order to achieve <50 copies/ml. If you don't achieve full suppression the authors report viral load rebounds quickly and resistance emerges.
AUTHOR DISCUSSION
Our results show that early treatment failure of a T-20-based salvage therapy regimen is associated with rapid emergence of T-20 resistance mutations in HR-1 of gp41. Variants carrying T-20 resistance mutations were detected at week 2, when the viral load was well below baseline levels (but not undetectable when resistance emerged; due to regimen that was not suppressive enough, not enough drugs patient was sensitive to), in samples from most subjects and became the predominant members of the viral quasispecies by week 4 in all 11 subjects. These results are consistent with previous studies of other groups10,17 but provide greater detail through clonal sequence analysis on the emergence of T-20 resistance mutations at time points soon after initiation of T-20-containing regimens. The earlier emergence of mutants with gp41 substitutions at amino acids 36 and 38 suggests that these mutants may have an initial fitness advantage over mutants with substitutions at codons 40 and 43, which tended to emerge later. It will be interesting to compare the relative fitness of these mutants by conducting growth competition experiments in the presence and absence of T-20.11
The prompt emergence of HR-1 mutations and their association with the rapid rebound of plasma HIV-1 RNA levels reflect the low genetic barrier to T-20 resistance and are consistent with the observations made with lamivudine and the nonnucleoside reverse transcriptase inhibitors (NNRTI).18,19 These results also suggest that resistance to T-20, as with lamivudine and the NNRTIs, can emerge rapidly in the setting of incomplete adherence or in patients interrupting therapy.
The results of clinical trials with novel protease inhibitors, such as tipranavir and darunavir, which retain activity against many highly drug-resistant HIV-1 isolates, demonstrate the important contribution of T-20 to achieving durable virologic suppression.20,21 The antiviral activity of these T-20-protease inhibitor combinations was greatest among subjects receiving T-20 for the first time, suggesting that the previous use of T-20 in a suboptimal regimen resulted in resistance that compromised its use in a subsequent regimen. Taken together, these results reinforce the importance of administering T-20 together with other active drugs to maximize the likelihood of suppressing plasma HIV-1 RNA levels to a level below the limits of detection (ie, <50 copies/mL).
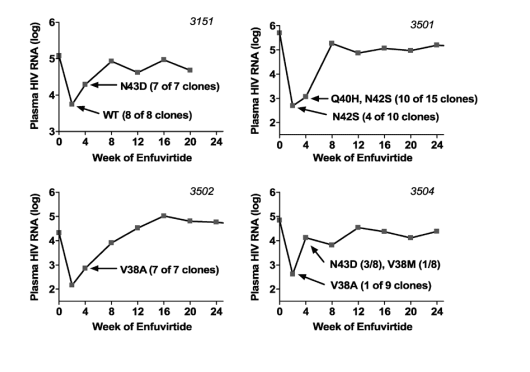
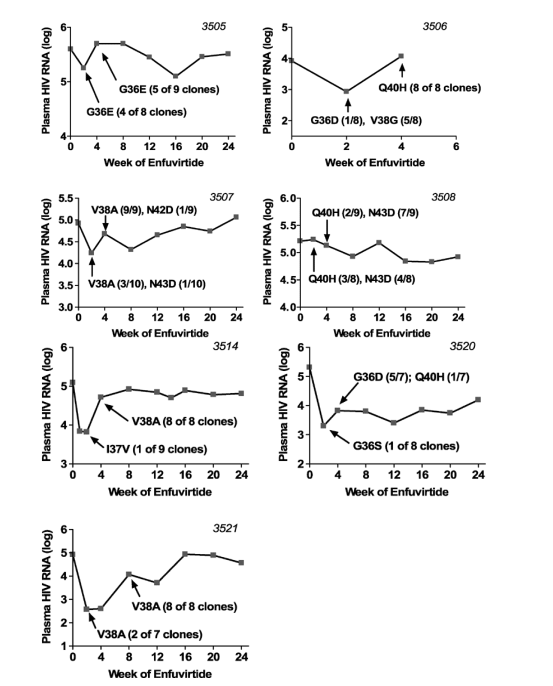
FIGURE 1. Plasma HIV-1 RNA levels over time for subjects initiating T-20 along with an optimized background regimen. [The continuous phenotypic susceptibility score15,16 of the background optimized regimen was 1.0 (IQR, 0.5-1.5). You can see in graphs of viral load declines below that patients had nice initial declines but rebounded within a few weeks & never reached <50 c/ml because evidently the regimens did not have enough sensitive background drugs]. Arrows indicate time of sampling for genotypic analysis. Amino acid substitutions occurring within positions 36 to 45 of the gp41 HR-1 domain are indicated.
Background
Enfuvirtide (T-20) is a synthetic 36-amino acid oligopeptide that inhibits fusion of HIV-1 to CD4+ target cells. Entry of HIV-1 requires the antiparallel association of 2 heptad repeats (HR-1 and HR-2) of the gp41 ectodomain to form a 6-helix bundle.1 Enfuvirtide binds to the trimeric HR-1 complex, thereby inhibiting fusion and blocking virus entry.2 The drug is highly effective in suppressing HIV-1 replication in treatment-experienced patients when combined with other active antiretroviral drugs.3-5
Resistance to T-20 is mediated by amino acid substitutions within HR-1 at amino acids 36 to 45 of gp41.6,7 The substitutions most frequently associated with T-20 resistance include G36D, G36S, G36V, or G36E, V38A, V38E, or V38M, Q40H, N42T, and N43D.8-10 These mutations confer significantly reduced binding of T-20 to HR-1 and a substantial decrease in antiviral activity in vitro.9 In addition, the N126K and S138A mutations in HR-2 may contribute to reduced T-20 susceptibility.8 Viruses carrying T-20 resistance mutations show reduced viral fitness in vitro in the absence of T-20.11
Previous reports of T-20 resistance generally have been based on the results of population sequencing of viral samples obtained from subjects receiving various doses of T-20 as monotherapy or added to a failing regimen, or after long-term T-20 therapy. To our knowledge, no study has focused on the early dynamics of T-20 resistance. Because the knowledge regarding the rate at which resistance emerges provides important insights into the genetic barrier to resistance, we performed a clonal analysis of HR-1 sequences from serial plasma samples obtained within the first month of treatment.
Abstract
Objectives: To study the dynamics of enfuvirtide (T-20) resistance development in HIV-1-infected subjects.
Patients and Methods: Clonal analysis of gp41 sequences was performed on serial samples obtained from HIV-1-infected subjects with early virologic failure of T-20-based regimens.
Results:
Enfuvirtide resistance mutations at codons 36 to 45 in the first heptad repeat of gp41 emerged within 2 weeks in most subjects and were associated with the return of plasma HIV-1 RNA level toward baseline by weeks 4 to 8. Mutations at codons 36 (G36E, G36D, or G36S) and 38 (V38A, V38G, or V38M) were the most commonly detected resistance mutations at week 2. Mutations at codons 40 (Q40H) and 43 (N43D) were more prevalent at week 4 than at week 2 and seemed to emerge more slowly than mutations at codons 36 and 38.
Conclusions: The rapid emergence of mutations associated with T-20 resistance in the absence of a fully suppressive antiretroviral regimen demonstrates a low genetic barrier to resistance and underscores the importance of combining T-20 with other active drugs when constructing regimens for highly treatment-experienced patients.
RESULTS
Cloning and sequencing of gp41 was successful in week 2 samples from 10 of 11 subjects with early failure of T-20-based therapy (the plasma HIV-1 RNA level in the week 2 sample from which gp41 could not be cloned was 146 copies/mL). The median CD4 count at the time of T-20 initiation was 8 cells/μL (interquartile range [IQR], 5-47 cells/μL), and the median plasma HIV-1 RNA level was 5.08 log10 copies/mL (IQR, 4.92-5.28 log10 copies/mL). The continuous phenotypic susceptibility score15,16 of the background optimized regimen was 1.0 (IQR, 0.5-1.5). Figure 1 shows the virologic response to therapy in these subjects. The median change in plasma HIV-1 RNA levels at weeks 2 and 4 was -1.73 (IQR, -2.19 to -0.98) and -0.58 (IQR, -1.64 to -0.31) log10 copies/mL, respectively. The median change in CD4 T-cell counts at weeks 2 and 4 were +29 (IQR, 10-41) and +26 (IQR, 14-46) cells/μL, respectively. In most cases, a 1-log10 or greater reduction in plasma HIV-1 RNA level at week 2 was followed promptly by rebound in plasma viremia.
Mutations at HR-1 positions associated with T-20 resistance were detected at week 2 in samples from 8 of 10 subjects (Fig. 2). Mutations at codon 38 (V38A, V38G, or V38M) were detected most often in these early samples (4 subjects each had at least one of these mutations). Three subjects had virus with mutations at codon 36 (G36E, G36D, or G36S). Mutations at codons 40 and 43 were detected in samples from only 1 and 2 subjects, respectively. In addition, a single clone with an I37V mutation was detected in 1 patient (patient 3514). In 1 subject (patient 3506), a mixture of mutations at codons 36 (G36D in 1 clone) and 38 (V38G in 5 clones) was found; these viruses were replaced at week 4 by a virus carrying the Q40H mutation. In a second subject (patient 3507), a mixture of V38A (3 clones) and N43D (1 clone) was present at week 2, but the V38A mutant predominated at week 4 (9/9 clones). A third subject (patient 3508) showed a mixture of Q40H (3 clones) and N43D (4 clones) mutants at week 2, which persisted at week 4 (6 and 3 clones, respectively).
No T-20-associated resistance mutations were detected in week 2 samples from 2 subjects with early treatment failure. In the case of subject 3151, all clones were wild type at codons 36 to 45 at week 2, but the N43D mutation was present in all 7 clones at week 4. In subject 3501, wild-type virus was replaced at week 2 by virus carrying the N42S polymorphism, which is not associated with T-20 resistance.7 Of note, the appearance of this polymorphism was accompanied by R46K and Q56R. At week 4, the Q40H resistance mutation was detected in 10 of 15 clones, along with persistence of N42S, R46K, and Q56R. Mutations in HR-1 outside the region of codons 36 to 45 that emerged on T-20 treatment included S35T, Q56R, and V72L. The emergence of Q56R seemed linked to S35T in clones from subject 3504, but the S35T mutation was present at baseline in clones from another subject (subject 3514), as was the V72L mutation (subject 3508).
METHODS
Study Population
Plasma samples were obtained from subjects enrolled in an ongoing prospective cohort study (Study of the Consequences of the Protease inhibitor Era [SCOPE]).12 All subjects provided written informed consent, and all aspects of this study were conducted according to institutional guidelines for experiments with human subjects. The subjects received T-20 (dosage, 90 mg 2 times a day) plus a background antiretroviral regimen selected on the basis of genotypic and phenotypic drug resistance testing (Monogram BioSciences, Inc, South San Francisco, CA). Thirty subjects enrolled in this study were followed up every 2 weeks for 4 weeks, and then every 4 weeks. From this cohort, we identified 11 subjects with evidence of early virologic failure, defined as lack of response or virologic rebound within the first 8 weeks of T-20 therapy. Samples from baseline and weeks 2 and 4 (or 8) were selected for detailed virologic analysis.
Cloning and Sequencing of HIV-1 gp41
Viral RNA was extracted from plasma using the QIAamp viral RNA Kit (Qiagen, Valencia, CA). A 650-base pair fragment of gp41 that included the HR-1 and HR-2 coding region (corresponding to nucleotides 7660-8310 of the Hxb2 sequence; information available at: http://hiv-web.lanl.gov ) was amplified by nested reverse transcriptase polymerase chain reaction (RT-PCR) using QIAgen OneStep RT-PCR kit with forward primer gp41OS 5' GAG GGA CAA TTG GAG AAG TGA ATT 3' and reverse primer gp41OA 5' GTG AGT ATC CCT GCC TAA CTC TAT 3' in the RT-PCR, and forward primer gp41IS 5' GGA GAA GTG AAT TAT ATA AAT ATA AAG 3' and reverse primer GP41A1 5' TTA AAC CTA CCA AGC CTC C 3' in the second-round PCR. After a reverse transcription reaction at 50 C for 30 minutes, reverse transcriptase was inactivated at 95 C for 15 minutes. The first-round PCR was performed using the following cycling conditions: 94 C for 30 seconds, 58 C for 30 seconds, and 72 C for 45 seconds for 35 cycles, followed by incubation at 72 C for 7 minutes. To minimize sampling bias, first-round RT-PCR products were performed in quadruplicate except for samples with plasma HIV-1 RNA level of less than 1000 copies/mL. (Performing quadruplicate RT-PCRs on the samples with low virus loads would have required additional plasma, which was not available to us. Although this approach might have introduced sampling bias, all but one of these samples from which we obtained sequence data showed presence of a mixture of wild-type and mutant sequences, suggesting that at least some of the heterogeneity present in the original sample had been preserved; Fig. 1). Five microliters of each quadruplicate first-round PCR product were pooled, and 3 μL of the mixture was then carried over to quadruplicate second-round PCR. The cycling conditions for the second-round PCR were 94 C for 30 seconds, 56 C for 30 seconds, and 72 C for 45 seconds for 35 cycles, followed by incubation at 72 C for 7 minutes.
|
|
| |
| |
|
|
|