| |
Radioactive antibodies hunt out HIV-infected cells
|
| |
| |
Antibodies tagged with radioactive elements might provide a new treatment.
Erika Check
Nature
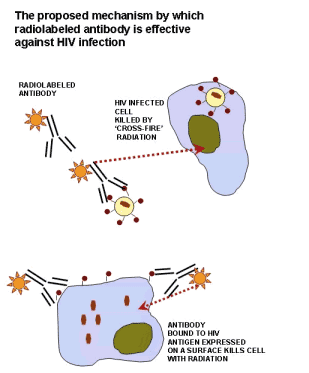
For decades researchers have wondered what it would take to eliminate the immunodeficiency virus HIV from a patient's body. Now they think that radioactive antibodies might do the trick. Scientists from the United States and Germany have combined antibodies that seek out cells infected with HIV with radioactive payloads that can destroy them, as they report in the 6 November issue of PLoS Medicine.
Harris Goldstein at the Albert Einstein College of Medicine in New York, who led the team, says the new approach might be used to kill HIV-infected cells soon after a person is infected, in an attempt to stop the virus establishing a permanent beachhead in the body. Such a treatment would probably be used in conjunction with the current drug treatment known as highly active antiretroviral therapy (HART). But it could also be used long after a person has been infected, perhaps together with therapeutic drugs - still in their experimental phase - that aim to flush HIV out of the redoubts in which it sequesters itself and make it more visible to the immune system.
"This is not a cure for HIV," Goldstein says. "This is a new concept for targeting HIV-infected cells and eliminating them."
Goldstein and his team tested their approach in cell cultures and in mice. First, the team constructed its weapons: radioactive chemicals joined to antibodies that recognize particular proteins displayed only on the surface of an HIV-infected cell. In tissue-culture experiments, the antibodies killed most of the HIV-infected cells.
The researchers then tested the radioactive antibodies in mice that had been specially engineered so that they contain human immune cells that HIC infects. The mice were infected with HIV and then injected with radiolabelled antibodies. The team found that the antibodies eliminated up to 99% of the HIV-infected cells from the mice, although the dose needed for almost total elimination was higher than that likely to be used in humans.
"This is a new concept for targeting HIV-infected cells and eliminating them."
Harris Goldstein,
Albert Einstein College of Medicine, New York.
Now, the team is trying to find a company that will test its approach in a clinical trial. Similar ideas have been tried before: toxins have been attached to antibodies, for example, in the hope that the toxins would destroy HIV-infected cells. That didn't work, but as radioactive antibodies are already used to treat certain cancers, the researchers hope that the new approach might also work for HIV.
"One advantage to this approach is that there's a significant body of work already in radioimmunotherapy for cancer cells, and this is sort of a cancer therapy approach to this problem, which may be what we need," says David Margolis, an HIV specialist at the University of North Carolina at Chapel Hill, who was not involved in the new work.
The main questions will be whether the new approach works in people, and whether it has toxic side effects. The main side effect is likely to be the accidental killing of cells not infected with HIV. The scientists behind this study said they didn't see evidence of toxicity in the mice, based on platelet counts in their blood, but only clinical trials will determine whether the same holds true in people.
Targeted Killing of Virally Infected Cells by Radiolabeled Antibodies to Viral Proteins
PLoS Medicine, Nov 7, 2006
Ekaterina Dadachova1*, Mahesh C. Patel1,2, Sima Toussi1, Christos Apostolidis3, Alfred Morgenstern3, Martin W. Brechbiel4, Miroslaw K. Gorny5, Susan Zolla-Pazner5,6, Arturo Casadevall1, Harris Goldstein1
1 Albert Einstein College of Medicine, Bronx, New York, United States of America, 2 Jacobi Medical Center, Bronx, New York, United States of America, 3 European Commission, Joint Research Centre, Institute for Transuranium Elements, Karlsruhe, Germany, 4 National Cancer Institute, National Institutes of Health, Bethesda, Maryland, United States of America, 5 New York University School of Medicine, New York, New York, United States of America, 6 Veterans Affairs New York Harbor Healthcare System, New York, New York, United States of America
Background
The HIV epidemic is a major threat to health in the developing and western worlds. A modality that targets and kills HIV-1-infected cells could have a major impact on the treatment of acute exposure and the elimination of persistent reservoirs of infected cells. The aim of this proof-of-principle study was to demonstrate the efficacy of a therapeutic strategy of targeting and eliminating HIV-1-infected cells with radiolabeled antibodies specific to viral proteins in vitro and in vivo.
Methods and Findings
Antibodies to HIV-1 envelope glycoproteins gp120 and gp41 labeled with radioisotopes bismuth 213 (213Bi) and rhenium 188 (188Re) selectively killed chronically HIV-1-infected human T cells and acutely HIV-1-infected human peripheral blood mononuclear cells (hPBMCs) in vitro. Treatment of severe combined immunodeficiency (SCID) mice harboring HIV-1-infected hPBMCs in their spleens with a 213Bi- or 188Re-labeled monoclonal antibody (mAb) to gp41 resulted in a 57% injected dose per gram uptake of radiolabeled mAb in the infected spleens and in a greater than 99% elimination of HIV-1-infected cells in a dose-dependent manner. The number of HIV-1-infected thymocytes decreased 2.5-fold in the human thymic implant grafts of SCID mice treated with the 188Re-labeled antibody to gp41 compared with those treated with the 188Re-control mAb. The treatment did not cause acute hematologic toxicity in the treated mice.
Conclusions
The current study demonstrates the effectiveness of HIV-targeted radioimmunotherapy and may provide a novel treatment option in combination with highly active antiretroviral therapy for the eradication of HIV.
Introduction
The HIV epidemic is a major threat to health in the developing and western worlds. Highly active antiretroviral therapy (HAART), a combination of drugs that inhibits enzymes essential for HIV-1 replication, can reduce the viremia and the onset of opportunistic infections in most patients and prolong survival [1]. However, HAART regimens are complicated and have significant toxicity [2-5]. Replication-competent virus that persists in infected cells emerges rapidly after the cessation of HAART [6-9]. The inability to eliminate HIV-1-infected cells also limits the time frame in which HAART initiated after exposure to HIV-1 can prevent infection [7]. Unless HAART is initiated within days of exposure, HIV infection of a nominal number of cells may be sufficient to develop sustained infection despite subsequent long-term HAART [10,11]. Once the clinical role of reservoir reduction is clarified, and under circumstances in which expected benefit outweighs safety considerations, a modality that targets and kills HIV-1-infected cells may become important for the treatment of acute exposure and elimination of persistent reservoirs of infected cells. To specifically kill HIV-1-infected cells, several approaches have been tried [12], including toxin-coupled antibodies that are directed at HIV-1-encoded envelope proteins and can eliminate infected cells [13,14], but none of these strategies has yet been shown to be effective in humans.
Radioimmunotherapy (RIT) takes advantage of the specificity of the antigen-antibody interaction to deliver lethal doses of radiation to target cells using radiolabeled antibodies [15]. Currently RIT is successfully used to treat of certain cancers. We demonstrated the usefulness of RIT for infectious diseases by treating murine cryptococcosis with a monoclonal antibody (mAb) to the Cryptococcus neoformans capsular glucuronoxylomannan labeled with bismuth 213 (213Bi) or rhenium 188 (188Re) [16], and extended the applicability of RIT to treatment of a bacterial infection [17]. We also recently suggested that RIT may be effective against chronically infected cells, including those with viral infections [18]. In contrast to RIT for fungal and bacterial diseases in which the target is the microbe, RIT for viral diseases targets the infected cells and consequently provides a general strategy for eliminating reservoirs of infected cells capable of producing infectious viruses. This approach could be particularly useful for treatment of drug-resistant HIV strains, which present an ever-increasing problem [19]. In the current proof-of-principle study, we examined the effectiveness of targeting and eliminating HIV-1-infected cells with radiolabeled antibodies to viral proteins.
Discussion
We have demonstrated that the antibodies to HIV-1 envelope glycoproteins gp120 and gp41 labeled with radioisotopes 213Bi and 188Re selectively killed chronically HIV-1-infected human T cells and acutely HIV-1-infected human peripheral blood mononuclear cells (hPBMCs) in vitro. Treatment of SCID mice harboring HIV-1-infected hPBMCs in their spleens with a 213Bi- or 188Re-labeled monoclonal antibody to gp41 viral protein resulted in a greater than 99% elimination of HIV-1-infected cells in a dose-dependent manner. The number of HIV-1-infected thymocytes decreased 2.5-fold in the human thymic implant grafts of SCID mice treated with the 188Re-labeled antibody to gp41 compared with those treated with the 188Re-control mAb. The treatment did not cause acute hematologic toxicity in the treated mice.
In cancer treatment, the success of FDA-approved drugs such as Zevalin and Bexxar (anti-CD20 mAbs labeled with yttrium 90 [90Y] and iodine 131 [131I], respectively) in the treatment of relapsed or refractory B cell non-Hodgkin lymphoma is proof of the enormous potential of RIT for targeted elimination of malignant cells and for treatment of patients who have failed all standard therapeutic regimens. Recent encouraging reports on the use of RIT as an initial treatment for follicular lymphoma [48] are making RIT a first-line anticancer therapy. This clinical experience using RIT creates a favorable environment for the introduction and use of RIT for treating HIV-1-infected patients. The effectiveness of RIT for HIV-1 infection is enhanced because the majority of long-lived infected cellular targets are lymphocytes, which are among the most radiosensitive cells in the body.
RIT of HIV-1 has five marked advantages over the immunotoxin approach. (1) The antibody used for radiation delivery does not need to be internalized to kill the cell. (2) Not every infected cell in the body needs to be targeted by the antibody because of the "cross-fire" effect. Consistent with this mechanism, 188Re-labeled mAbs may be more effective in vivo than in vitro (Figures 1A and 3A). In vitro, the "cross-fire" radiation is largely deposited on the two-dimensional surface represented by the cell layer at the bottom of a tissue culture well, whereas in vivo there are many infected cells in the nearby three-dimensional space such that "cross-fire" radiation is more effective. In this regard, beta-emitting radionuclides, which kill cells preferentially by "cross-fire" [15], have been successfully used for therapy of leukemia, a single-cell disease [49], and in preclinical RIT of systemic fungal infections [16,39,50]. (3) In contrast to immunotoxins, the radioisotope linked to the antibody is unlikely to elicit significant immune responses that would limit subsequent use. (4) RIT is potentially less toxic than immunotoxins, because the chemistry of linking different radioisotopes including 188Re and 213Bi to the antibodies has been well developed, and the exceptional stability of radiolabeled mAbs in vitro and in vivo has been confirmed [15,42,51]. (5) The availability of many isotopes differing in half-life and radiation type [15] offers great versatility for designing RIT. Nevertheless, combination therapy consisting of RIT and immunotoxins may provide greater advantages than either modality alone.
We have recently shown that RIT of infection works through the combination of cell-killing mechanisms such as "direct hit" and "cross-fire," promotion of apoptotic death of targeted cells, generation of reactive oxygen species and modulation of the inflammatory response [50]. All of these mechanisms require certain levels of radioactivity to manifest themselves. It is likely that in our study the threshold dose at which those mechanisms become effective in killing HIV-infected cells is in the range 40-80 (1.48-2.96 MBq) μCi. There are numerous published data on the existence of threshold RIT doses below which no therapeutic effects on tumors in animals are observed and above which the effects quickly reach maximum [42, 52]. However, determination of the threshold doses in RIT is not considered to be of practical importance as in the clinic this therapy is administered as maximum tolerated dose (MTD) to maximize the therapeutic effect. These results establish that RIT can effectively target and kill HIV-1-infected hPBMCs in vivo.
Based on the data accumulated in clinical RIT of cancer, the primary toxic effect of RIT applied to infections is likely to be bone marrow suppression. Important determinants of the extent and duration of myelosuppression include bone marrow reserve (based on prior cytotoxic therapy and extent of disease involvement), total infection burden, and spleen size [53,54]. Nevertheless, RIT of non-Hodgkin lymphoma is effective in patients who have received several unsuccessful courses of chemotherapy and consequently have depleted bone marrow reserves and weakened immunity-not unlike some patients with advanced HIV-1 infection. The application of RIT to infectious diseases will require optimization of the dose to ascertain and minimize toxic effects.
In addition, when using a radioactive therapy in patients there is always a concern of long-term effects such as the possible subsequent development of neoplasms arising from radiation-induced mutations. However, this risk should be extremely low after short-term exposure and would likely be outweighed by the benefits of treating or preventing HIV infections [55]. It is also extremely unlikely that particulate radiation utilized in RIT will cause mutation of the HIV virus, the genomic information of which is carried by two positive RNA strands and not by DNA, and ionizing radiation is a weak mutagen compared to chemical mutagens [56]. Furthermore, HIV replication already has an inherently high rate of viral mutation.
Acutely exposed individuals lack HIV-specific antibodies that could reduce the effectiveness of RIT by competitively binding to gp41, so RIT could be used for treatment initiated after HIV exposure. However, the effectiveness of treatment in patients with established HIV infection may be reduced by competition between the radionuclide-linked antibody and the patient's own antiviral antibodies directed to the same antigen on HIV-1-infected cells. If this competition occurs, it could be circumvented by increasing the RIT antibody dosage or by coupling the radionuclides to very high affinity antibodies likely to bind more effectively to the gp41 than self-antibodies. The latter approach would be enhanced by using cocktails of mAbs against noncompeting epitopes and, following the well-known radiation oncology approach of dose fractionation, each antibody in the antibody cocktail would carry a partial dose (one-half or one-third, depending on the composition of the cocktail). Our group is currently investigating the role of competition between radiolabeled mAb 246-D and cold polyclonal human anti-gp41 antibody binding to HIV-1 gp41 and killing HIV-1-infected cells.
If advanced to clinical use, RIT might be most effective when used in combination with HAART [57], which blocks virus replication in newly infected cells. The major therapeutic use of RIT will likely be as an adjunct with HAART to prevent HIV infection when administered to individuals within the first days of exposure to HIV. Even if infection is not prevented in some cases, initial treatment of patients soon after infection may reduce the initial number of HIV-1-infected cells and thereby reduce viral set point. RIT also may be a useful adjunct for protocols designed to "flush out" quiescent, latently infected lymphocytes by the administration of factors, such as valproic acid, that promote HIV replication [58]. Moreover, since viral resistance is often a result of mutations introduced during replication, a therapy that reduces the number of HIV-producing cells may reduce the emergence of drug-resistant strains. To avoid selection of cells harboring antibody-resistant viruses, a cocktail of mAbs to various epitopes on the HIV envelope may be desirable. We envision that the availability of RIT could provide a novel treatment option that may hasten the day when curative regimens are available for the eradication of HIV-1 infection.
Results
To determine the ability of RIT to kill HIV-1-infected cells, goat polyclonal antibodies to gp120 were labeled with radioisotopes with distinctly different emission characteristics-213Bi (a radioisotope that emits alpha particles, which are He atoms with a charge of +2 and mass of 4) and 188Re (a radioisotope that emits high-energy beta particles [electrons]). 213Bi and 188Re have different emission ranges in tissue: 50-80 μm for 213Bi versus 10 mm (on average) for 188Re. Both radioisotopes have been used in preclinical and clinical settings [29-31]; we used both to investigate if HIV-1-infected cells can be killed with either alpha- (213Bi) or beta-radiation (188Re).
The chronically HIV-1-infected ACH-2 cells were incubated with 188Re-anti-gp120, 188Re-18B7 (irrelevant murine mAb), or cold anti-gp120. Substantial killing of ACH-2 cells with 188Re-anti-gp120 polyclonal antibody was observed compared to the minimal effect of the control Ab linked to 188Re with the same specific radioactivity or with cold anti-gp120 Ab (Figure 1A). Elimination of the ACH-2 cells was not complete. By virtue of their mass, charge, and energy the relative biological effectiveness of alpha particles is significantly higher than that of beta particles emitted by 188Re [32,33]. Therefore, we examined the effectiveness of the anti-gp120 linked to the alpha particle emitter 213Bi for killing HIV-1-infected cells. Almost all of the ACH-2 cells were killed by 5 μCi (0.185 MBq) of 213Bi-anti-gp120 compared to the minimal cytocidal activity of an irrelevant mAb linked to 213Bi or cold anti-gp120 Ab (Figure 1B).
Because the HIV envelope protein gp120 is expressed extracellularly, it was the initial target for antibody-directed killing of HIV-infected cells. However, toxins coupled to gp41-specific antibodies were reported to be more effective for in vivo elimination of HIV-1-infected cells than were toxins linked to anti-gp120 [14,26,34]. An advantage of targeting gp41 over gp120 with antibodies is that gp41 is a transmembrane protein that is reliably expressed on the surface of infected cells, is not readily shed, and has a highly conserved sequence [35]. Therefore, we examined the ability of radioisotopes coupled to a human mAb specific for gp41 to target and kill HIV-1-infected hPBMCs in vitro and in vivo. For these studies, we used the human anti-gp41 mAb 246-D, which binds specifically to HIV-1-infected cells as demonstrated by flow cytometry of hPBMCs infected with HIV-1JR-CSF (Figure 2A). 246-D does not bind to the six-helix bundle that forms after gp120 CD4 intervention, but rather to an epitope, near the disulfide loop of gp41, that is an immunodominant epitope [36] and is broadly cross-reactive with genetically diverse HIV-1 strains [37,38]. When 246-D was linked with 213Bi, it was very effective in eliminating HIV-1-infected hPBMCs when compared to cold 246-D and the irrelevant mAb 1418 labeled with 213Bi (Figure 2B). We observed that more hPBMCs were killed by 213Bi-246-D than overall percentage of infected cells in the sample. This most likely reflects a "cross-fire" effect from alpha particles emanating from adjacent HIV-1-infected cells that contained bound 213Bi-mAb in the setting of cell crowding at the bottom of tissue culture wells. Importantly, in the absence of HIV-1 antigens, incubation of noninfected hPBMCs with 213Bi-246-D produced no significant killing of the cultured cells (Figure 2B), and no killing of noninfected hPBMCs was observed post-treatment with 188Re-246-D as well (unpublished data).
We next examined the in vivo ability of RIT with radiolabeled 246-D to eliminate HIV-1-infected cells. For these studies, hPBMCs infected with HIV-1JR-CSF were injected into the spleens of SCID mice and the mice were treated as indicated. We used a 80 μCi (2.96 MBq) dose for 188Re-246-D and 100 μCi (3.7 MBq) for 213Bi-246-D, because these doses were therapeutic and safe in experimental RIT of fungal and bacterial infections [17,39]. The mice were evaluated 72 h later for the presence of residual HIV-1-infected cells by quantitative coculture [25]. The 72 h time period was chosen to give the sufficient time for the 188Re-labeled mAb to deliver a lethal dose of radioactivity to the cells, as 188Re half-life is 16.9 h and several half-lives are required for a given radionuclide to deliver the dose to the target. Treatment of mice with 188Re-labeled mAb 246-D administered either before or after intrasplenic injection with HIV-1JR-CSF-infected hPBMCs dramatically reduced the subsequent number of HIV-1-infected cells (p = 0.016 and p = 0.008, respectively). Infected cells were also killed after treatment of mice with 213Bi-246-D (Figure 3A) (p = 0.008). In contrast, the administration of the matching amounts of cold 246-D (p = 1.0) or of a radioisotope-coupled irrelevant control mAb did not reduce the average number of infected cells in the SCID mouse spleens (p = 1.0 and p = 0.06 for 188Re- and 213Bi-labeled control mAb, respectively). It is likely that 188Re-246-D was more effective in vivo than 213Bi-246-D due to the longer physical half-life of 188Re (16.9 h versus 46 min), which allowed the labeled mAbs to reach infected cells while still carrying a high-activity "payload."
To investigate the dose-response effect, the mice were treated with 40 and 160 μCi of 188Re-246-D, corresponding to 50% and 200% of the therapeutic dose, respectively. While 40 μCi (1.48 MBq) 188Re-246-D was not effective in killing HIV-infected hPBMCs in vivo (p = 1.0), 160 μCi (5.92 MBq) dose almost completely eliminated the HIV-infected cells (p = 0.03) (Figure 3B).
To further investigate the specificity of radiolabeled mAb binding to gp41 on HIV-infected hPBMCs we compared the splenic uptake of 188Re-246-D in mice injected intrasplenically with noninfected hPBMCs and HIV-1-infected hPBMCs. The uptake expressed as percentage of ID/g of spleen (with standard deviation) was 8% ± 4% and 57% ± 10% ID/g for noninfected and infected hPBMCs, respectively. This result indicates that the 188Re-246-D was targeted in vivo to HIV-1-infected cells.
We evaluated the hematological toxicity of radiolabeled 246-D during HIV-1 infection in the SCID mice by platelet counts. The platelet count nadir usually occurs 1 wk after radiolabeled antibody administration to tumor-bearing animals [40,41]. We did not observe any changes in platelet counts in mice treated with 100 μCi (3.7 MBq) of 213Bi-246-D on days 4, 8, and 15 post-treatment compared to nontreated infected controls; platelet counts were stable at 1.5 X 109 (± 0.2 X 109) platelets/ml of blood (unpublished data). For mice given 160 μCi of 188Re-246-D-the highest dose used in this study-a slight nonsignificant drop in platelet count was noted on day 7 post-treatment, but counts returned to normal by day 15 (Figure 4). This lack of hematologic toxicity likely reflects the very specific targeting of HIV-1-infected hPBMCs by radiolabeled mAb, since gp41 antigen is expressed only on infected cells in the mouse. In fact, one of the advantages of using RIT to treat infections as opposed to cancer is that, in contrast to tumor cells, cells expressing microbial antigens are antigenically very different from host tissues and thus provide the potential for exquisite specificity and low cross-reactivity. This safety profile is compatible with our previous data showing that RIT was also relatively nontoxic in mouse models of experimental fungal and bacterial infection [17,39]. A large therapeutic window is available because the therapeutic effect was achieved with activities significantly lower than reported maximum tolerated activity for 188Re-labeled (800 μCi (29.6 MBq) for intravenous injection [42]) and 213Bi-labeled IgGs (in excess of 1,000 μCi (37 MBq) when given IP [42]). In this regard, IP administration of the radiolabeled mAbs utilized in our study has proved to be better tolerated than the intravenous route [42], and it helps to circumvent the problem of often relatively low specific activity of radiolabeled mAbs (in mCi/ml) by allowing the administration of higher volumes than during intravenous administration.
To further examine the in vivo capacity of RIT to target and eliminate HIV-1-infected lymphocytes, we utilized our well-characterized thy/liv-SCID-hu mouse model [43-45]. This system uses SCID mice implanted under their kidney capsules with fetal human thymic and liver tissue that develops into a structure closely resembling a human thymus; this thymic implant is readily infectible with clinical isolates of HIV-1. This mouse model can be used to evaluate the in vivo capacity of candidate anti-HIV agents to inhibit HIV-1 infection [43,45,46]. Initially we measured the biodistribution of IP-injected 188Re-246-D in thy/liv-SCID-hu mice with HIV-infected and noninfected thymic implants (Table 1). The uptake of 246-D in infected implants was higher than in noninfected implants; however, the overall uptake in the infected implants 18 h postinjection was modest, most likely due to the relatively poor vascularization of the implants. We have previously demonstrated that HIV-1-infected thy/liv-SCID-hu mice develop HIV plasma viremia, which may correlate with the higher uptake of 246-D in the blood as well as spleen, liver, and kidneys of the HIV-infected mice.
Because uptake of the radiolabeled anti-gp41 mAb in infected implants of thy/liv-SCID-hu was modest after IP injection, the therapy experiments were performed using intraimplant injections of radiolabeled mAbs to accommodate the predominantly murine vasculature of the implant, which may contribute to decreased transport of the radiolabeled antibody. Sufficient blood supply to the tissue treated with RIT is crucial for successful treatment, as it has been established that early in RIT development antibodies mostly localize along the blood vessels [47]. As shown in Figure 5A, there was a 2.5-fold reduction in the number of HIV-1-infected thymocytes in the human thymic implant grafts of mice treated with 188Re-246-D compared to those treated with the 188Re-control mAb. To directly confirm the anti-HIV effect of the 188Re-anti-gp41 treatment, we compared the level of infection in the human thymic implants of a biopsy taken before treatment with the degree of infection after treatment. The mean number of HIV-infected thymocytes was reduced more than 95% by 188Re-anti-gp41 in three of the four treated mice (Figure 5B). Taken together, these results demonstrate that RIT using 188Re-anti-gp41 mAb potently and selectively eliminates HIV-1-infected cells.
Methods
Antibodies
Goat polyclonal antibodies against gp120 (IgG) used in preliminary in vitro experiments was purchased from Biodesign International (Saco, Maine, United States). Murine 18B7 mAb (IgG1) to C. neoformans [20] was used as an irrelevant control. Human mAb to gp41, 246-D (cluster I), was produced as described [21]. Human mAb 1418 (IgG1) to parvovirus B19 [22] was used as an irrelevant control for mAb 246-D, and human mAb 447 (IgG3) to the V3 loop of HIV-1 gp120 [23] was used as a positive control in the FACS studies. Antibodies were purified by protein A or G chromatography prior to labeling.
Radioisotopes and Radiolabeling of the Antibodies
Antibodies were labeled with alpha-emitter 213Bi (half-life 45.6 min) obtained from 225Ac generator as described in [24] or with beta-emitter 188Re (half-life 16.9 h) as described in [16].
In Vitro Killing of ACH-2 Cells and Human PBMCs with Radiolabeled mAbs
The ACH-2 cell line is a latent T cell clone infected with HIV-IIIB that produces steady low levels of viruses that is markedly increased by stimulation with phorbol myristate; it was obtained through the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program (Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH: ACH-2, #349, from Dr. Thomas Folks). At 48 h after stimulation with phorbol myristate, ACH-2 cells cultured in triplicate wells were treated with 0-50 μCi (0-1.85 MBq) of 188Re-labeled anti-gp120 or the irrelevant control mAb 18B7; or 0-20 μCi (0-0.74 MBq) of 213Bi-labeled anti-gp120 or mAb 18B7 or with matching amounts (2.5-12.5 μg) of "cold" (unlabeled) anti-gp120 mAb. Approximately 2 X 105 cells were used for each condition. The cells were incubated with radiolabeled or cold mAbs at 37 C for 3 h, transferred into fresh cell culture medium, and then incubated in 5% CO2 at 37 C for 72 h. The number of viable cells was then assessed by Trypan blue dye exclusion assay.
The effect of radiolabeled HIV-1 glycoprotein-binding mAbs on HIV-1-infected cells was studied in vitro using human peripheral blood mononuclear cells (hPBMCs) obtained from New York Blood Center (New York, New York, United States). hPBMCs were stimulated with phytohemagglutinin (PHA) and interleukin-2 for 48 h and then infected with HIV-1 strain JR-CSF (NIH AIDS Research and Reference Reagent Program, National Institute of Allergy and Infectious Diseases, NIH: HIV-1JR-CSF, #394, from Dr. Irvin S. Y. Chen). Although the number of ACH-2 cells infected with HIV-1 was almost 100%, only a fraction (∼10%-30%) of the hPBMCs were infected with HIV-1 as determined by limiting dilution coculture technique [25]. Subsequently in the text we refer to the cells exposed to HIV-1 as "infected" cells and those that were not exposed to the virus as "noninfected" cells. At 48 h after infection, infected cells cultured in triplicate wells were treated with 213Bi-labeled mAb 246-D, or 213Bi-labeled irrelevant control mAb 1418, or matching amounts of cold 246-D, as above. To verify that 246-D did not bind to the surface of noninfected hPBMCs, noninfected cells were subjected to treatment with 213Bi-246-D or 188Re-246-D. The number of viable cells was then assessed by Trypan blue dye exclusion assay.
Flow Cytometric Analysis of mAbs Binding to Virus-Infected Cells
Binding studies of human mAbs to the surface of hPBMCs infected with HIV-1JR-CSF were performed as described previously [26]. Briefly, PHA-stimulated hPBMCs were infected with 1 ml of stock HIV-1JR-CSF virus and cultured for 13 d in medium supplemented with human recombinant interleukin-2 (20 U/ml; Boehringer Mannheim Biochemicals, Indianapolis, Indiana, United States). The cells in duplicate wells were incubated with each human mAb at 10 μg/ml for 1 h on ice, washed and reincubated with phycoerythrin-labeled goat F(ab��)2 anti-human IgG(γ) (Caltag Laboratories, Burlingame, California, United States). Using a FACScan flow cytometer (Becton Dickinson, Palo Alto, California, United States), live lymphocytes were selected for analysis by gating with forward and 90 scatter. The negative control consisted of cells from infected cultures stained with the conjugated anti-IgG in the absence of a human mAb.
Treatment of HIV-1 Infection in Intrasplenically Infected SCID Mice with Radiolabeled mAbs
The procedures used in animal studies described below were reviewed and approved by the Albert Einstein College of Medicine Animal Institute Committee. These experiments utilized a mouse model; first, PHA-activated hPBMCs were infected with HIV-1JR-CSF as described above, then 48 h later these infected hPBMCs were injected intrasplenically (25 X 106 cells per mouse) into SCID mice. Mice were treated intraperitoneally 1 h later with one of the following: 20 μg of cold 246-D (n = 4); 100 μCi (3.7 MBq) (20 μg) of 213Bi-1418 (n = 5) or 80 μCi (2.96 MBq) (20 μg) of 188Re-1418 (n = 3) as isotype-matching controls; or 80 μCi (2.96 MBq) (20 μg) of 188Re-246-D (n = 4) or 100 μCi (3.7 MBq) (20 μg) of 213Bi-246-D (n = 5). In one experiment mice were given 80 μCi (2.96 MBq) (20 μg) of 188Re-246-D intraperitoneally (IP) 1 h prior to infection with infected hPBMCs (n = 4). In dose-response experiments mice were injected IP with 188Re-246-D (20 μg) with activity of 40 μCi (1.48 MBq) (n = 4), with 160 μCi (5.92 MBq) (n = 4), with 20 μg of cold 246-D (n = 4), or with no treatment (n = 4). Mice were sacrificed and spleens were harvested and processed 72 h postinjection. The number of HIV-1-infected cells present in the spleen was measured using limiting dilution quantitative coculture as described [25]. This technique measures the number of cells capable of producing infectious HIV-1.
Cells isolated from each spleen were diluted 5-fold (in the range 1 X 106 to 3.2 X 102 cells) and cultured in duplicate at 37 C in 24-well culture plates with PHA-activated hPBMCs (1 X 106 cells) in 2.0 ml of RPMI 1640 medium containing fetal calf serum (10% vol/vol) and interleukin-2 (32 U/ml). The HIV-1 p24 antigen content of the supernatant was measured 1 wk later, using the HIV-1 p24 core profile ELISA (DuPont-NEN, Boston, Massachusetts, United States). The lowest number of added cells that infected at least half the duplicate cultures with HIV-1 was determined; this number represented the frequency of cells productively infected with HIV-1 in each spleen, reported as the tissue culture-infecting dose causing 50% infection (TCID50)/106 splenocytes. For example, if coculture of 106 cells was positive and coculture of 2 X 105 cells was negative, we assigned this a value of 1 TCID50/106 cells because 106 cells was the lowest number of cells that generated productive infection.
Determination of Splenic Uptake of Radiolabeled mAbs
Two groups of SCID mice (four mice per group) were used in this experiment. One group was injected intrasplenically with HIV-1-infected hPBMCs (25 X 106 cells/ mouse) and another with noninfected hPBMCs (20 X 106 cells/ mouse) as described above. After 1 h, 20 μCi (0.74 MBq) (20 μg) of 188Re-246-D was given IP to each mouse, and at 3 h postinjection the animals were sacrificed, their spleens removed and weighed, radioactivity counted in a gamma counter, and the percentage of injected dose per gram (ID/g) calculated.
Determination of Platelet Counts in Mice Treated with Radiolabeled mAbs
Platelet counts were used as a marker of RIT toxicity in treated animals. For measurement of platelet counts, the blood of SCID mice (five mice per group) injected intrasplenically with HIV-1-infected hPBMCs and either treated with 100 μCi (3.7 MBq) (20 μg) of 213Bi-246-D or with 160 μCi (5.92 MBq) (20 μg) of 188Re-246-D IP 1 h after treatment with infected hPBMCs or untreated was collected from the tail vein into 200 μl of 1% ammonium oxalate on days 0, 3, 8, and 15 days post-therapy. Platelets were counted in a hemocytometer, using phase contrast, at 400X magnification, as described in [27].
Construction of thy/liv-SCID-hu Mice and Titration of HIV-1-Infected Mononuclear Cells in the hu-thy/liv Implant by Limiting Dilution Coculture
The thy/liv-SCID-hu mice. were constructed by implanting SCID mice (6-8 wk old) with human fetal thymic and liver tissue under the kidney capsules of SCID mice as described [28]. Briefly, after SCID mice were anesthetized with pentobarbital (40-80 mg/kg), approximately ten pieces of syngeneic human fetal thymic and liver tissue were implanted under the kidney capsules, and 3 mo later the size of implanted tissue had increased by more than 20-fold.
Biodistribution of mAb 188Re-246-D in thy/liv-SCID-hu Mice
Three thy/liv-SCID-hu mice were infected with HIV-1JR-CSF via its thymic implant, as described above. Only one implant in each mouse (each mouse had two implants) was infected. Three thy/liv-SCID-hu mice with noninfected implants were used as controls. At 8 d postinfection mice were injected IP with 188Re-246-D (60 μCi (2.22 MBq), 20 μg of mAb) and sacrificed to assay for biodistribution 18 h later. Only the infected implant was taken out to check biodistribution. The blood and major organs were removed, weighed, and counted in a gamma counter, and the percentage of ID/g was calculated.
Treatment of HIV-1 Infection in Infected thy/liv-SCID-hu Mice with Radiolabeled mAbs
Thy/liv-SCID-hu mice were infected by direct injection of HIV-1JR-CSF (800 TCID50) into the human thymic implant. Selected implants were biopsied 1 wk later, and the number of HIV-1-infected thymocytes was quantified by limiting dilution coculture. One group of mice (four mice) was treated with 188Re-246-D (160 μCi (5.92 MBq), 20 μg mAb) and another group (four mice) with the same activity of 188Re-1418. After 72 h, the mice were sacrificed and the number of HIV-1-infected thymocytes was determined by quantitative coculture.
Statistical Analysis
The statistical aspects of data analysis were discussed with Dr. Y. Lo, a statistician at the Einstein/Montefiore Medical Center, Center for AIDS Research. The sample sizes in animal experiments were preplanned taking into consideration availability of animals. The observers were fully blinded to the active or control nature of the experimental arm being assayed. The differences between the numbers of TCID50/106 splenocytes for differently treated groups during in vivo therapy studies were analyzed by nonparametric Mann-Whitney test using Prism software (GraphPad, San Diego, California, United States). Differences were considered statistically significant when
|
|
| |
| |
|
|
|