| |
The Management of Treatment-Experienced HIV-Infected Patients: New Drugs and Drug Combinations
|
| |
| |
Clinical Infectious Diseases Jan 15 2009;48:214-221
Lucy E. Wilson and Joel E. Gallant
Division of Infectious Diseases, Johns Hopkins School of Medicine, Baltimore, Maryland
"recent availability of new antiretroviral agents for HIV treatment....has led to a revision of current treatment guidelines, which now state that the goal of antiretroviral therapy in all patients is suppression of the plasma HIV RNA level to <50 copies/mL.....it is critical that clinicians use the new agents carefully and that patients understand the importance of adherence. The DUET [4, 5], MOTIVATE [20], and BENCHMRK [12-14] studies suggest that an effective and durable regimen should include at least 2-and preferably 3-active agents. The years 2007 and 2008 will be viewed as a landmark in this history of HIV therapy, because for the first time, almost all patients with access to therapy can now achieve virological suppression. Whether this is the beginning of a new era or just a brief "honeymoon period" will depend on how we use these valuable new agents".
The recent availability of new antiretroviral agents for the treatment of human immunodeficiency virus (HIV) infection has increased treatment options and has improved the durability, tolerability, and long-term efficacy of antiretroviral therapy, even among patients with extensive treatment experience and high levels of drug resistance. This expansion of therapeutic options has led to a revision of current treatment guidelines, which now state that the goal of antiretroviral therapy in all patients is suppression of the plasma HIV RNA level to <50 copies/mL. Successful management of infection for treatment-experienced patients with the new agents requires an understanding of their pharmacology and resistance patterns and the appropriate use of laboratory testing to optimize regimen selection. This review discusses the use of recently approved antiretroviral agents in the management of HIV infection in treatment-experienced patients.
New Agents, New Goals
The recent approval of new antiretroviral agents within existing and novel classes has ushered in a transformation of the HIV treatment landscape. Treatment-experienced patients with extensive drug resistance, who previously had "untreatable virus," now have multiple options for suppressive therapy. Treatment guidelines from the Department of Health and Human Services and the International AIDS Society-USA now state that the goal of therapy is virological suppression to <50 copies/mL for all patients, not just those undergoing initial therapy [1, 2].
Although these recent developments represent a major step forward, they also create new challenges for clinicians, who must now be knowledgeable about the use and interpretation of a variety of types of resistance and tropism assays, who must understand complex issues of pharmacokinetics and drug interactions, and who must keep abreast of the expanding treatment armamentarium. Expert care is essential to avoid the rapid emergence of resistance to the new agents.
In the pre-HAART era and in the early years of the HAART era, there were many causes of treatment failure. However, because of the potency and efficacy of current therapies, failure of initial therapy that was chosen on the basis of drug resistance test results is usually attributable to nonadherence; uncommon exceptions are transmitted nonnucleoside reverse-transcriptase inhibitor (NNRTI) resistance not detectable by baseline genotype and negative pharmacokinetic interactions, including food effects. For all others, the levels of efficacy and durability of an appropriately chosen regimen should be high.
New Antiretroviral Agents
NNRTIs
Etravirine, an NNRTI that has activity against virus with NNRTI resistance, received US Food and Drug Administration (FDA) approval in January 2008. Approval was based primarily on the results of DUET 1 and 2 [3-6], ongoing 96-week, double-blind clinical trials in which 612 subjects (pooled sample size from 2 identical studies) were treated with etravirine or placebo in combination with an optimized background regimen (OBR) that included darunavir-ritonavir plus nucleoside reverse-transcriptase inhibitors (NRTIs) with or without enfuvirtide. Pooled 48-week data indicated that 60% of those treated with etravirine had viral loads <50 copies/mL, compared with 40% of patients who received placebo (p<.001, by intention-to-treat-time to loss of virological response analysis), with improved efficacy in those with more active drugs in the OBR.
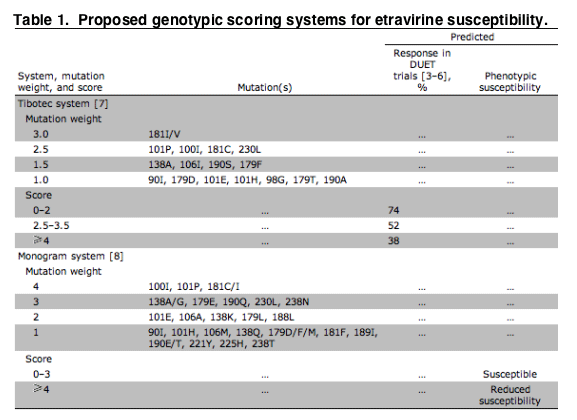
The most common adverse effects associated with etravirine are rash and nausea, although only rash occurred more frequently than did other adverse effects for patients who received placebo (22% vs. 11%) [4, 5]. Rash was mild and self-limited, and it infrequently required drug discontinuation. Etravirine remains effective in patients with common NNRTI mutations, including K103N, which does not affect drug susceptibility. Baseline mutations associated with a poorer response to etravirine in the DUET trials were 90I, 98G, 100I, 101E/P, 106I, 179D/F, 181C/I/V, and 190A/S; etravirine had limited activity in patients with >3 of these mutations. Two weighted genotype-scoring systems for etravirine susceptibility have been proposed-namely, one by Tibotec [7], in which the genotype is used to estimate response in the DUET trials, the other by Monogram Biosciences [8], in which the genotype is used to estimate phenotypic susceptibility (table 1). Because failure of other NNRTIs can lead to etravirine cross-resistance, it is important to discontinue the use of "first-generation" NNRTIs in patients without complete virological suppression [9]. In patients with prior NNRTI use, genotypes drawn at the time of NNRTI failure may be more accurate than current genotypes. However, not all recognized etravirine mutations were assessed in older commercial genotypes. Phenotype assays now measure etravirine susceptibility, although they may not be helpful in patients with virological failure who are no longer taking NNRTIs.
There are currently no data on the use of etravirine for treatment-naive patients; however, etravirine could be considered among those infected with NNRTI-resistant virus. The need for twice-daily dosing and the current pill burden also reduce the appeal of etravirine as a first-line agent, although once-daily dosing is being evaluated on the basis of the long half-life of etravirine.
Because it induces CYP3A4, etravirine is not currently recommended for use in combination with protease inhibitors (PIs), except darunavir-ritonavir, saquinavir-ritonavir, and possibly lopinavir-ritonavir [10]. Concomitant use of phenytoin and rifampin is contraindicated. When etravirine is combined with maraviroc, dosing adjustments are required (table 2).

Integrase Inhibitors
Integrase inhibitors block the strand transfer step of the integration process-that is, the insertion of reverse-transcribed viral DNA into host DNA. FDA approval of raltegravir was based on the BENCHMRK 1 and 2 studies [12, 13], in which highly treatment-experienced patients were randomized to receive raltegravir or placebo plus OBR. In BENCHMRK 1, 65% of those who received raltegravir achieved virological suppression (HIV load, <50 copies/mL) at 48 weeks, compared with 31% of those who received placebo (p<.001) [12]. In BENCHMRK 2, the results were 60% and 34%, respectively [13]. Complete viral suppression with raltegravir treatment occurred most frequently when the OBR included >1 other active drug.
Raltegravir is dosed as a single tablet given twice daily and has a favorable adverse effect profile [14, 15]. The most common adverse reactions in clinical trials were diarrhea and nausea, although these did not occur more frequently than with placebo.
Resistance to raltegravir is typically associated with mutations at either Q148 or N155, in combination with other mutations in the integrase gene [16]. Phenotypic resistance testing for integrase inhibitors is now commercially available. However, there is currently only 1 integrase inhibitor available, and it is expected that there will be cross-resistance between raltegravir and elvitegravir, an integrase inhibitor in development.
Raltegravir is indicated for treatment-experienced patients who experience failure with other regimens. Raltegravir should be combined with at least 1 and preferably 2 other active agents, to avoid resistance. Raltegravir is also being used as a replacement for suppressive agents that cause toxicity (e.g., enfuvirtide). Initial data support its use in first-line therapy (in combination with 2 NRTIs) [14], although data from phase III trials will be needed before this strategy can be recommended.
CCR5 Antagonists
In August 2007, maraviroc became the first FDA-approved CCR5 antagonist. Drugs in this class attach to the CCR5 coreceptor on the CD4 cell surface, blocking entry of R5 virus, which uses that coreceptor to enter the cell. R5 virus is most prevalent in early disease and is the most likely virus to be transmitted; with time, tropism can shift with emergence of X4 virus, which uses the CXCR4 coreceptor. X4 virus is usually present in combination with R5 virus, and dual-tropic virus (virus that can make use of both coreceptors) also exists. Tropism assays cannot distinguish between dual-tropic virus and mixed populations, which are collectively referred to as dual/mixed (DM) virus. Tropism shifts have been associated with accelerated decrease in CD4 cell count and accelerated disease progression [17], although the causal relationship is unclear.
Maraviroc is administered twice daily in combination with other active agents to patients assumed, on the basis of pretreatment tropism, to be infected with pure R5 virus. The dose varies depending on the other drugs in the antiretroviral regimen (table 3) [19].

Approval of maraviroc was based on the MOTIVATE 1 and 2 studies, which randomized >1000 experienced patients with R5 virus to receive 1 of 2 doses of maraviroc versus placebo in combination with OBR20. At 48 weeks, 43%-45% of those receiving maraviroc had viral loads <50 copies/mL, compared with 17% receiving placebo, as determined by intention-to-treat analysis of the pooled study data. Efficacy in all study arms was associated with the number of active drugs in the OBR. No important safety differences were found between the maraviroc and placebo arms in the first 48 weeks of study.
Baseline tropism screening indicated that 56% of subjects had R5 virus; 44% had DM or X4 virus and were excluded from participation in this study [21]. Of those who enrolled with R5 virus, 8% were subsequently found to have DM virus on the first day of treatment; they were more likely than those without DM virus to experience virological failure [20]. This apparent discrepancy does not reflect a true tropism shift; instead, it reflects the lower sensitivity of the original tropism assay (Trofile; Monogram Biosciences [22]) for detection of DM virus when the virus comprised a minority of the viral population. The new, enhanced Trofile ES sensitivity assay is more accurate in detecting DM or X4 virus [23]. With the original assay, selection of preexisting DM or X4 virus was the most common cause of early failure of maraviroc in treatment-experienced patients [24]. Patients who experience early failure with R5 virus infection have low drug levels, presumably because of nonadherence to treatment [25]. Failure to treat R5 virus infection can also occur because of mutations in the V3 loop [26]; however, it is not yet possible to measure genotypic or phenotypic resistance with use of commercially available assays.
Maraviroc is indicated for treatment-experienced patients. Treatment-naive patients with higher CD4 cell counts are more likely to have R5 virus than are treatment-experienced patients or those with more-advanced disease. However, maraviroc did not meet criteria for noninferiority compared with efavirenz in treatment-naive patients in the MERIT trial [27].(from Jules: at the recent ICAAC meeting, 9/08, a reanalysis of the MERT study using the newer sensitive Tropism assay found maraviroc was not inferior to efavirenz). In addition, the need for twice-daily dosing, the requirement for baseline tropism testing, and the lack of long-term safety data for this class, with a novel and immune-based mechanism of action, make it unlikely that this agent will be used in treatment-naive patients in the near future.
Because tropism testing requires a viral load of >1000 copies/mL, it is not recommended that maraviroc be substituted for other agents in a suppressive regimen. It is unclear whether tropism should be reassessed in patients who experience therapy failure with maraviroc [20, 28]. Detection of DM or X4 virus predicts little virological benefit to additional therapy with a CCR5 antagonist. However, there is evidence of a short-term increase in the CD4 cell count even when CCR5 antagonists are given to patients with DM virus infection [20, 29]. This finding is reassuring, because there were initial safety concerns that inadvertent selection of X4 virus could result in more-rapid decrease in CD4 cell count. An increase in CD4 cell count could prove beneficial in patients without other options; however, durability and long-term consequences of this selection pressure have not been studied. The possibility of a CD4 cell count increase is not an argument for routine use of these agents without baseline tropism testing. The goal of therapy is maximal virological suppression; use of a CCR5 antagonist in a patient with DM virus infection provides inadequate support for the other drugs in the regimen and increases the chance of resistance.
PIs
It is now recommended that all PIs be boosted with ritonavir. Failure of PI-based regimens in patients without preexisting PI resistance is rarely associated with the emergence of new PI mutations. However, extensive PI resistance is still seen in patients who took unboosted PIs in the earlier years of the HAART era, especially when PI treatment was continued despite virological failure. Many patients with broad PI cross-resistance may benefit from the more recently approved PIs darunavir and tipranavir.
Darunavir. Darunavir received FDA approval in June 2006 for use with ritonavir in treatment-experienced patients. When administered at a dosage of 600 mg (1 tablet) twice daily with 100 mg of ritonavir, it is active against many strains of PI-resistant virus [30]. In the POWER studies [31], treatment-experienced patients who received darunavir-ritonavir plus OBR had better virological responses at 48 weeks than did those who received OBR alone. Decreased virological response was seen with 3 of the following mutations: 11I, 32I, 33F, 47V, 50V, 54L/M, 74P, 76V, 84V, and 89V. Optimal response to darunavir-ritonavir was associated with a phenotypic change of 10-fold [30, 31].
The TITAN study compared darunavir-ritonavir with lopinavir-ritonavir plus OBR in treatment-experienced subjects who were naive to both agents [32]. At 48 weeks, virological response was superior in the darunavir-ritonavir arm. When those with baseline phenotypic resistance to lopinavir were excluded from the analysis, the difference between treatment arms was no longer significant, although there was still a trend favoring darunavir-ritonavir. There were more virological failures and the emergence of new PI mutations among subjects taking lopinavir-ritonavir, who were also more likely to lose susceptibility to other PIs [32].
Darunavir is now indicated for both treatment-experienced and treatment-naive patients. In the ARTEMIS trial, a lower dose of darunavir-ritonavir (800/100 mg once daily) was at least as effective as lopinavir-ritonavir in treatment-naive patients, and a 400-mg tablet was recently released.
Toxicity is similar to that of other PIs [31, 33], although darunavir-ritonavir was associated with less diarrhea and more rash, compared with lopinavir-ritonavir, in the TITAN study. The FDA recently issued a warning that was based on postmarketing reports with regard to increased hepatitis among individuals with preexisting liver dysfunction [34]. However, during clinical trials, significant hepatotoxicity was uncommon, even among HIV-infected patients coinfected with hepatatis C virus or hepatatis B virus who received darunavir-ritonavir.
Tipranavir. Tipranavir was approved by the FDA in 2005 for use with ritonavir in treatment-experienced patients with extensive PI resistance. Approval was based on the RESIST trials [35], which demonstrated superior virological response among treatment-experienced patients taking tipranavir-ritonavir versus comparator PIs at 48 and 96 weeks. A mutation score can be calculated by adding the number of the following PI resistance mutations: 10V, 13V, 20M/R/V, 33F, 35G, 36I, 43T, 46L, 47V, 54A/M/V, 58E, 69K, 74P, 82L/T, 83D, and 84V. Optimal tipranavir-ritonavir response is indicated by a score of 0-1, intermediate response is indicated by a score of 2-7, and minimal response is indicated by a score >8 [35]. Phenotype assays may be more predictive of tipranavir activity, as they were in the POWER trials with darunavir [30, 31].
In the RESIST trial [35], tipranavir-ritonavir caused more hyperlipidemia and hepatotoxicity than did comparator PIs, which may be because of the need for a higher boosting dose of ritonavir (200 mg twice daily). Because tipranavir can increase serious bleeding risk, including intracranial hemorrhage, caution is advised for patients who have had recent surgery, previous head injury, or bleeding diatheses or who are receiving medications that increase bleeding risk [36]. Tipranavir cannot be combined with PIs other than ritonavir, because tipranavir induces CYP3A4 metabolism and lowers PI plasma concentrations.
Resistance testing is critical for PI selection in a treatment-experienced patient. Although susceptibility can be assessed genotypically, phenotype or virtual phenotype testing may be preferable in patients with multiple PI mutations, because they allow for quantification of partial susceptibility and comparison among agents. When there is no clear resistance advantage of one drug over the other, darunavir is typically preferred because of its more favorable toxicity profile.
Fusion Inhibitors
The availability and tolerability of the new agents has substantially decreased enfuvirtide use. The TORO studies [37, 38] demonstrated the efficacy and durability of enfuvirtide in treatment-experienced patients, and POWER [30, 31], RESIST [35], MOTIVATE [20], and BENCHMRK [12-14] demonstrated the benefit of enfuvirtide when combined with new agents. However, enfuvirtide is expensive, requires time-consuming reconstitution and twice-daily injection, and causes painful injection-site reactions. Enfuvirtide is an option for treatment-experienced patients with DM or X4 virus infection and cross-resistance to darunavir, tipranavir, and/or etravirine or after failure of the new agents. (from Jules: I'm not so sure I agree to so easily reject consideration of Fuzeon. Some highly-treatment experienced patients may have so much PI and NNRTI resistance that consideration of maraviroc and/or Fuzeon may be in order; I suggest caution so you can protect raltegravir).
NRTIs
The role of NRTIs in treatment-experienced patients is somewhat unclear. NRTIs are often used in treatment of patients with resistance, given persistent antiretroviral activity despite multiple NRTI mutations. However, the new drugs have made it possible to use 3 fully active agents in many patients, which brings into question the benefit of continued NRTI therapy. ACTG 5241 is an ongoing study designed to address this question; in the meantime, the decision about whether to include NRTIs should depend on resistance testing, the number of other active drugs available, and evidence of NRTI toxicity.
Many patients with extensive NRTI resistance have taken multiple nonsuppressive thymidine analogue-containing regimens. The trend away from use of thymidine analogues in treatment-naive patients may decrease development of extensive NRTI resistance and may improve future NRTI sequencing options. In addition, there are novel NRTIs in development that show evidence of activity against NRTI resistance virus.
Using Laboratory Tests to Guide Treatment Decisions
Resistance testing. Selection of the optimal regimen for treatment-experienced patients requires an understanding of available laboratory tests. Approaches to resistance testing include genotype assays, phenotype assays, and the virtual phenotype assay VircoTYPE [39], in which a genotype is used to estimate the phenotype on the basis of algorithms derived from a database of samples with paired genotypes and phenotypes. Interpretation of genotype results can be complex when multiple mutations are present. Numerous algorithms are used that vary in terms of the weight they give to specific mutations. Phenotype or virtual phenotype assays may have advantages in treatment-experienced patients, especially for selection of PIs. Previous resistance tests are as important as current ones, because mutations that emerged during earlier therapy are archived and remain clinically important but may not be detected if a drug in the relevant class is no longer being taken. If prior genotype analyses were not performed or are not available, it may be necessary to make assumptions about resistance on the basis of treatment history. It is now possible to test for resistance to raltegravir by phenotype, but it is not possible to test for resistance to maraviroc, as discussed above in CCR5 Antagonists.
Tropism assays. Tropism testing is essential before starting maraviroc, which should not be used in patients with X4-tropic or DM-tropic virus infection. The new Trofile ES assay has improved sensitivity for detection of low-level X4 or DM virus infection [23, 40, 41]. Efforts to develop lower-cost genotypic assays for tropism are under way, but the tests developed thus far are not sufficiently sensitive [42].
Conclusions
The multitude of new options and the strength of existing drug combinations (table 4) give most patients excellent prospects for achieving virological suppression. However, the drug pipeline holds little short-term promise for patients whose virus cannot be fully suppressed with available agents. Rilpivirine (TMC278), an NNRTI that is being studied in treatment-naive patients, is unlikely to be effective against etravirine-resistant virus [6]. Patients with raltegravir-resistant virus infection are likely to have cross-resistance to elvitegravir [16, 43-45]. Vicriviroc, a CCR5 antagonist, will be inactive among those ineligible for maraviroc because of DM or X4 virus infection.
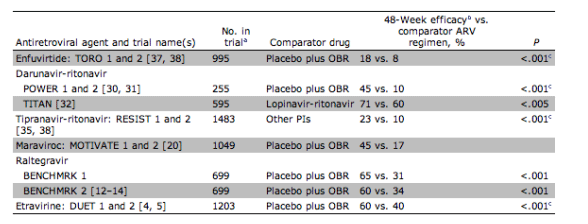
NOTE. Cross-study comparisons should be made with caution, because clinical trials vary with respect to enrollment criteria, patient population, time period, and agents allowed in optimized background regimen (OBR).
aIntention to treat.
bTo achieve a viral load <50 copies/mL in the intention-to-treat analysis.
cCombined data.
There are some investigational agents, such as TNX355, an anti-CD4 monoclonal antibody, and bevirimat, a maturation inhibitor, that have novel mechanisms of action and will be active in patients with resistance to the current agents, but the future of these agents remains uncertain, and they will not be available soon. Thus, it is critical that clinicians use the new agents carefully and that patients understand the importance of adherence. The DUET [4, 5], MOTIVATE [20], and BENCHMRK [12-14] studies suggest that an effective and durable regimen should include at least 2-and preferably 3-active agents. The years 2007 and 2008 will be viewed as a landmark in this history of HIV therapy, because for the first time, almost all patients with access to therapy can now achieve virological suppression. Whether this is the beginning of a new era or just a brief "honeymoon period" will depend on how we use these valuable new agents.
|
|
| |
| |
|
|
|