| |
Important FDA Label changes to Viramune (nevirapine) oral solution and tablets
|
| |
| |
On June 24, 2008, FDA approved labeling changes to the Viramune (nevirapine) oral solution and tablets to reflect various updates, including:
- Dosing recommendations for pediatric patients 15 days to 2 months of age.
- Dose recommendations for all pediatric age groups are now based on body surface area (BSA) instead of weight-based dosing. Studies were conducted comparing weight-based dosing and BSA-based dosing. While comparable drug concentrations are achieved with either method, BSA dosing allows for smoother dose transitions between pediatric age groups and is therefore preferred.
- Addition of data from a pharmacokinetic hepatic impairment study
- Revision of the recommendation that nevirapine not be administered to patients with severe hepatic impairment to a recommendation that nevirapine not be administered to patients with moderate (Childs Pugh B) or severe (Childs Pugh C) hepatic impairment.
- Revision of recommendations for the occurrence of rash during the once daily lead-in phase of dosing. The label now states that lead-in dosing should not be extended beyond 28 days of dosing.
Important changes made to the product label include the following:
Under Dosage and Administrations (2.0)
Pediatric Patients (2.2)
The recommended oral dose for pediatric patients 15 days and older is 150 mg/m2 once daily for 14 days followed by 150 mg/m2 twice daily thereafter. The total daily dose should not exceed 400 mg for any patient.
Table 1 Calculation of the Volume of VIRAMUNE Oral Suspension (50 mg/5 mL) Required for Pediatric Dosing Based on Body Surface and a Dose of 150 mg/m2
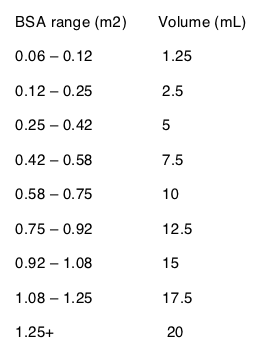
Dosage Adjustment (2.4)
Patients with Rash
VIRAMUNE should be discontinued if a patient experiences severe rash or any rash accompanied by constitutional findings [see Boxed Warning, Warnings and Precautions (5.2), and Patient Counseling Information (17.1)]. A patient experiencing mild to moderate rash without constitutional symptoms during the 14-day lead-in period of 200 mg/day (150 mg/m2/day in pediatric patients) should not have their VIRAMUNE dose increased until the rash has resolved [see Warnings and Precautions (5.2) and Patient Counseling Information (17.1)]. The total duration of the once daily lead-in dosing period should not exceed 28 days at which point an alternative regimen should be sought.
Under Use in Specific Populations (8.0)
Hepatic Impairment (8.7)
Because increased nevirapine levels and nevirapine accumulation may be observed in patients with serious liver disease, do not administer VIRAMUNE to patients with moderate or severe (Child Pugh Class B or C, respectively) hepatic impairment [see Contraindications (4), Warnings and Precautions (5.1), and Clinical Pharmacology (12.3)].
Under Pharmacokinetics (12.3)
Hepatic Impairment
In a steady state study comparing 46 patients with mild (n=17; expansion of some portal areas; Ishak Score 1-2), moderate (n=20; expansion of most portal areas with occasional portal-to-portal and portal-to-central bridging; Ishak Score 3-4), or severe (n=9; marked bridging with occasional cirrhosis without decompensation indicating Child-Pugh A; Ishak Score 5-6) fibrosis as a measure of hepatic impairment, the multiple dose pharmacokinetic disposition of nevirapine and its five oxidative metabolites were not altered. However, approximately 15% of these patients with hepatic fibrosis had nevirapine trough concentrations above 9,000 ug/mL (2-fold the usual mean trough). Therefore, patients with hepatic impairment should be monitored carefully for evidence of drug induced toxicity [see Warnings and Precautions (5.1)]. The patients studied were receiving antiretroviral therapy containing Viramune 200 mg twice-daily for at least 6 weeks prior to pharmacokinetic sampling, with a median duration of therapy of 3.4 years.
Pediatric Patients
Pharmacokinetic data for nevirapine have been derived from two sources: a 48 week pediatric trial in South Africa (BI Trial 1100.1368) involving 123 HIV-1 positive, antiretroviral naive patients aged 3 months to 16 years; and a consolidated analysis of five Pediatric AIDS Clinical Trials Group (PACTG) protocols comprising 495 patients aged 14 days to 19 years.
BI Trial 1100.1368 studied the safety, efficacy, and pharmacokinetics of a weight-based and a body surface area (BSA)-based dosing regimen of nevirapine. In the weight-based regimen, pediatric patients up to 8 years of age received a dose of 4 mg/kg once daily for two weeks followed by 7 mg/kg twice daily thereafter. Patients 8 years and older were dosed 4 mg/kg once daily for two weeks followed by 4 mg/kg twice daily thereafter. In the BSA regimen all pediatric patients received 150 mg/m2 once daily for two weeks followed by 150 mg/m2 twice daily thereafter [see Use in Specific Populations (8.4) and Adverse Reactions (6.2)]. Dosing of nevirapine at 150 mg/m2 BID (after a two-week lead in of 150 mg/m2 QD) produced geometric mean or mean trough nevirapine concentrations between 4-6 ā╩g/mL (as targeted from adult data). In addition, the observed trough nevirapine concentrations were comparable between the two dosing regimens studied (BSA and weight-based methods).
The consolidated analysis of Pediatric AIDS Clinical Trials Group (PACTG) protocols 245, 356, 366, 377, and 403 allowed for the evaluation of pediatric patients less than 3 months of age (n=17). The plasma nevirapine concentrations observed were within the range observed in adults and the remainder of the pediatric population, but were more variable between patients, particularly in the second month of age. For dose recommendations for pediatric patients see Dosage and Administration (2.2).
Under Clinical Studies (14.0)
Clinical Studies in Pediatric Patients (14.2)
The pediatric safety and efficacy of VIRAMUNE was examined in BI Trial 1100.1368, an open-label, randomized clinical study performed inSouth Africa in which 123 HIV-1 infected treatment naive patients between 3 months and 16 years of age received VIRAMUNE oral solution for 48 weeks. Patients were divided into 4 age groups (3 months to <2 years, 2 to <7 years, 7 to <12 years, and 12 to üģ16 years) and randomized to receive one of two VIRAMUNE doses, determined by 2 different dosing methods [body surface area (150mg/m2) and weight-based dosing (4 or 7mg/kg)] in combination with zidovudine and lamivudine [see Adverse Reactions (6.2),Use in Specific Population (8.4), and Clinical Pharmacology (12.3)]. The total daily dose of VIRAMUNE did not exceed 400 mg in either regimen. There were 66 patients in the body surface area (BSA) dosing group and 57 patients in the weight-based (BW) dosing group.
Baseline demographics included: 49% male; 81% Black and 19% Caucasian; 4% had previous exposure to ARVs. Patients had a median baseline HIV RNA of 5.45 log10 copies/mL and a median baseline CD4 cell count of 527 cells/mm3 (range 37-2279). One hundred and five (85%) completed the 48 weeks period while 18 (15%) discontinued prematurely. Of the patients who discontinued prematurely, 9 (7%) discontinued due to adverse reactions and 3 (2%) discontinued due to virologic failure. Overall the proportion of patients who achieved and maintained an HIV RNA <400 copies/mL at 48 weeks was 47% (58/123). For dose recommendations for pediatric patients see Dosage and Administration (2.2).
You can view the complete, updated labeling using this link to
Drugs@FDA(http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.SearchAction&SearchTerm=Viramune&SearchType=BasicSearch), clicking on the appropriate dosage form, and folloing links for ügLabeling Information."
Richard Klein _Office of Special Health Issues _Food and Drug Administration
Kimberly Struble _Division of Antiviral Drug Products _Food and Drug Administration
|
|
| |
| |
|
|
|