| |
ARTEMIS Study: Efficacy and safety of once-daily darunavir/ritonavir versus lopinavir/ritonavir in treatment-naive HIV-1-infected patients at week 48
|
| |
| |
AIDS:Volume 22(12)31 July 2008p 1389-1397
Ortiz, Robertoa; DeJesus, Edwina; Khanlou, Homayoonb; Voronin, Evgeniyc; van Lunzen, Jand; Andrade-Villanueva, Jaimee; Fourie, Janf; De Meyer, Sandrag; De Pauw, Martineg; Lefebvre, Erich; Vangeneugden, Tonyg; Spinosa-Guzman, Sabrinag
aOrlando Immunology Center, Orlando, Florida, USA
bAIDS Healthcare Foundation, Los Angeles, California, USA
cRepublican Hospital of Infectious Diseases Clinical AIDS Center, St Petersburg, Russia
dInfectious Diseases Unit, University Medical Centre Hamburg-Eppendorf, Germany
eHospital Civil de Guadalajara, Guadalajara, Mexico
fChronic Diseases of Lifestyle Unit, Tygerberg, South Africa
gTibotec BVBA, Mechelen, Belgium
hTibotec Department, Janssen-Cilag B.V., Tilburg, Netherlands.
Abstract
Background: The present primary analysis of AntiRetroviral Therapy with TMC114 ExaMined In naive Subjects (ARTEMIS) compares the efficacy and safety of once-daily darunavir/ritonavir (DRV/r) with that of lopinavir/ritonavir (LPV/r) in treatment-naive patients.
Methods: Patients with HIV-1 RNA at least 5000 copies/ml were stratified by HIV-1 RNA and CD4 cell count in a phase III, open-label trial, and randomized to receive DRV/r 800/100 mg qd or LPV/r 800/200 mg total daily dose (bid or qd) plus fixed-dose tenofovir and emtricitabine for 192 weeks. The primary objective was to demonstrate non-inferiority of DRV/r as compared with LPV/r in HIV-1 RNA less than 50 copies/ml per-protocol time-to-loss of virologic response at 48 weeks.
Results: Six hundred and eighty-nine patients were randomized and treated; mean baseline HIV-1 RNA: 4.85 log10 copies/ml and median CD4 count: 225 cells/μl. At 48 weeks, 84% of DRV/r and 78% of LPV/r patients achieved HIV-1 RNA less than 50 copies/ml (estimated difference = 5.6 [95% confidence interval -0.1-11]%), demonstrating non-inferiority of DRV/r as compared with LPV/r (P < 0.001; per-protocol time-to-loss of virologic response). Patients with HIV-1 RNA at least 100 000 copies/ml had a significantly higher response rate with DRV/r (79%) versus LPV/r (67%; P < 0.05). Median CD4 cell count increases (non-completer = failure; cells/μl) were 137 for DRV/r and 141 for LPV/r. DRV/r had a lower incidence of possibly treatment-related grade 2-4 gastrointestinal-related adverse events (7 versus 14%) and treatment-related moderate-to-severe diarrhea (4 versus 10%) than LPV/r. Adverse events leading to discontinuation were DRV/r: 3% and LPV/r: 7%.
Conclusion: DRV/r 800/100 mg qd was non-inferior to LPV/r 800/200 mg at 48 weeks, with a more favorable safety profile. Significantly higher response rates were observed with DRV/r in patients with HIV-1 RNA at least 100 000 copies/ml. DRV/r 800/100 mg offers a new effective and well tolerated once-daily, first-line treatment option for treatment-naive patients.
Introduction
First-line antiretroviral therapy (ART) provides the greatest opportunity to fully suppress HIV replication and prevent the emergence of drug-resistant strains that lead to treatment failure and compromise future drug treatment strategies. An initial ART regimen should be potent, durable, able to prevent or delay the onset of drug resistance and should also have good tolerability and a convenient dose schedule.
Darunavir (DRV; TMC114) with low-dose ritonavir at a dose of 600/100 mg bid has demonstrated sustained efficacy and favorable safety in patients with a broad range of treatment experience [1-3]. On the basis of the results from the Performance Of TMC114/r When Evaluated in treatment-experienced patients with PI Resistance (POWER) studies in treatment-experienced patients [4,5], once-daily darunavir/ritonavir (DRV/r) 800/100 mg was selected for patients with no previous treatment experience. The suitability of once-daily dosing in this population is supported by the long half-life of DRV in the presence of ritonavir (15 h) [6].
Here we present the 48-week primary analysis of ARTEMIS (AntiRetroviral Therapy with TMC114 ExaMined In naive Subjects), a study assessing the efficacy and safety of DRV/r 800/100 mg qd as compared with lopinavir/ritonavir (LPV/r) in treatment-naive HIV-1-infected patients over 192 weeks.
Methods
Study design
ARTEMIS is an ongoing, randomized, phase III, open-label trial comparing the efficacy, safety, and tolerability of once-daily DRV/r as compared with LPV/r in treatment-naive patients. This study is being conducted in centers across 26 countries. The trial comprises a screening period of 2-4 weeks and a treatment period of 192 weeks (and a 96-week rollover phase). Patients were stratified by plasma HIV-1 RNA (<100 000, ≥100 000 copies/ml) and CD4 cell count (<200, ≥200 cells/μl) at screening, and then randomized in a 1: 1 ratio to receive either DRV/r 800/100 mg qd (darunavir 400 mg tablet not yet available) or LPV/r 800/200 mg total daily dose (400/100 mg bid or 800/200 mg qd depending on local regulatory approval and investigator or patient preference or both). Randomization was performed by means of a predefined randomization list, using a central randomization system (interactive voice response) to ensure balance across treatments groups in each stratum (stratification described above). Initially LPV/r was given as soft-gel capsules, but subject to availability and local approval, patients were switched to LPV/r tablets. Patients receiving LPV/r qd with regimen intolerance could switch to bid dosing. In addition, all patients received a fixed background regimen, tenofovir (300 mg qd) and emtricitabine (200 mg qd).
The primary objective of the trial was to demonstrate non-inferiority of DRV/r 800/100 mg qd as compared with LPV/r 800/200 mg total daily dose in virologic response defined as a confirmed plasma HIV-1 RNA less than 50 copies/ml by per-protocol time-to-loss of virologic response (PP-TLOVR) at 48 weeks. Secondary objectives included evaluation of other virologic and immunologic parameters over 192 weeks (including proportion of patients with HIV-1 RNA less than 400 copies/ml, change in HIV-1 RNA and CD4 cell count change from baseline); evaluation of safety and tolerability; and in the event of non-inferiority, testing for superiority of DRV/r over LPV/r (planned analysis). The primary analysis described here was performed when all patients had been treated for 48 weeks or had discontinued earlier.
Study population
ARTEMIS inclusion criteria included treatment-naive HIV-1-infected patients aged at least 18 years, with plasma HIV-1 RNA at least 5000 copies/ml. Patients with the following conditions were excluded: active AIDS-defining illness; any clinically significant disease; clinical or laboratory evidence of significantly decreased hepatic function or decompensation; acute viral hepatitis at screening or calculated creatinine clearance less than 70 ml/min. Individuals with primary HIV infection or those pregnant or breastfeeding were also excluded. Patients with grade 3 or 4 laboratory abnormalities (Division of AIDS grading table) were not eligible with some exceptions (diabetes or asymptomatic glucose, triglyceride or cholesterol elevations) unless clinical assessment identified health risks. Patients coinfected with chronic hepatitis B or C were allowed entry if their condition was clinically stable and they did not require treatment during the study period.
Written informed consent was obtained from all patients. Study protocols were reviewed and approved by the appropriate institutional ethics committees and health authorities and were conducted in accordance with the Declaration of Helsinki.
Study evaluations and statistical methods
The study commenced on 28 September 2005. Data up to the cutoff date of 13 June 2007 are included. Efficacy and safety variables were assessed at screening, baseline, and at each visit (at 2 weeks, then every 4 weeks until week 16, at week 24, and every 12 weeks until week 192). The per-protocol population (DRV/r, N = 340 and LPV/r, N = 346; all randomized patients who had received study medication and had not taken disallowed therapy for more than 1 week) was used to test for non-inferiority.
TLOVR algorithm was used to assess virologic response (HIV-1 RNA < 400 and <50 copies/ml). If non-inferiority was established, superiority testing would be performed in the intent-to-treat (ITT) population (DRV/r N = 343 and LPV/r N = 346; all randomized and treated patients). Confirmed virologic responses (<50 copies/ml) at week 48 were compared between DRV/r and LPV/r, using the TLOVR algorithm: if at week 48, the lower limit of the 95% two-sided confidence interval (CI) of the difference between DRV/r and LPV/r was higher than -12%, non-inferiority of DRV/r over LPV/r could be concluded. The predefined delta of -12% was considered appropriate as it was small relative to observed differences between LPV/r and other active regimens in previous studies [7]. A total of 330 patients in each treatment arm were needed to provide adequate statistical power to ascertain whether DRV/r was non-inferior to LPV/r, with a one-sided significance level of �� = 0.025 and 90% power to detect any difference. Statistical comparisons were made using a logistic regression model (corrected for stratification factors). A ��2 test was used to compare safety results and efficacy results across baseline HIV-1 RNA and CD4 cell count strata. Viral phenotypic and genotypic assessments were performed by Virco BVBA (Mechelen, Belgium), by means of the Antivirogram and VircoTYPE HIV-1 assays, respectively. For resistance determination, virologic failure was identified using the TLOVR non-virologic failure censored algorithm where the responses at timepoints after discontinuation were not imputed for patients who discontinued due to reasons other than virologic failure (patients who discontinued before week 12 were excluded from the virologic failure determination). Patients with virologic failure were defined as those whose virus was never suppressed to HIV-1 RNA less than 50 copies/ml after at least 24 weeks of treatment or those who achieved that level of response but rebounded (rebound was defined as confirmed on-study HIV-1 RNA at least 50 copies/ml, or last observed HIV-1 RNA at least 50 copies/ml followed by discontinuation).
Patients were asked to complete a modified medication adherence self-report inventory (M-MASRI) questionnaire and report their adherence to treatment over time. Adherence rates were transformed to a binary variable using a 95% cutoff to define adherence [8].
The ITT population was used for the safety analysis. Assessments were performed on the basis of adverse event data (classified according to the Division of AIDS table), clinical laboratory tests (hematology, coagulation testing, biochemistry, hepatitis serology/viremia and urinalysis), cardiovascular variables (vital signs and electrocardiograph), physical examination, and anthropometric measurements. An independent Data and Safety Monitoring Board reviewed the data.
Sparse blood sampling was performed for 335 patients randomized to the DRV/r arm to predict trough concentration for DRV using a population pharmacokinetic model.
Results
Patient disposition and baseline characteristics
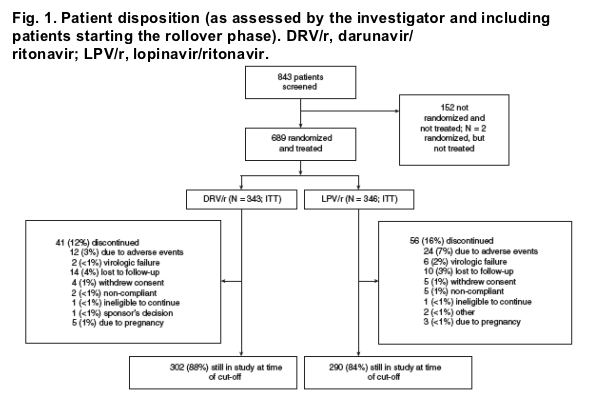
Of the 843 patients screened, 689 were randomized and treated (Fig. 1). All DRV/r patients received DRV/r qd. At the primary analysis cutoff timepoint, LPV/r dosing/formulation was as follows: 77% received bid, 15% qd, and 8% received both bid and qd; 15% of patients received soft-gel capsules, 2% tablets and 83% switched from capsules to tablets. The overall discontinuation rate was relatively low in both treatment arms (DRV/r 12%, LPV/r 16%). Most discontinuations were due to adverse events (DRV/r 3%, LPV/r 7%); 2% of LPV/r and less than 1% of DRV/r patients discontinued due to virologic failure.
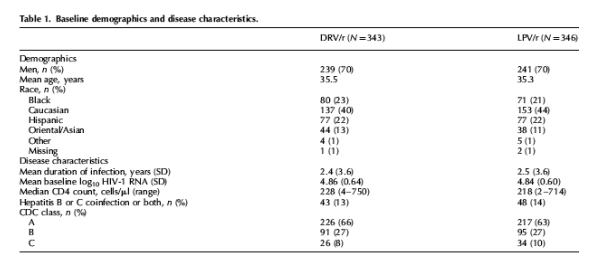
Baseline characteristics were well balanced between treatment arms, stratification factors and countries (Table 1). The trial included a diverse population where women and non-Caucasians were well represented (30 and 58%, respectively). Consistent with the naive ART status of patients, few patients (9%) had Centers for Disease Control category C HIV infection. At baseline, 34% of patients had at least 100 000 copies/ml and median CD4 count was 225 cells/μl.
Efficacy
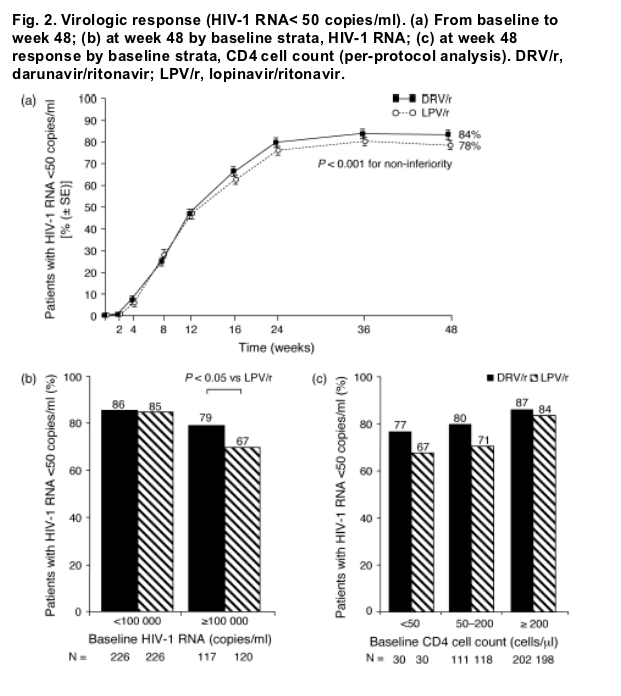
At week 48, 84% of DRV/r and 78% of LPV/r patients had a confirmed virologic response of HIV-1 RNA less than 50 copies/ml in the per-protocol population (TLOVR; Fig. 2a). The ITT results were identical to those of the per-protocol population. On the basis of the model, the estimated difference between treatment responses was 5.6% (95% CI, -0.1-11): the lower limit of the 95% CI was greater than -12% (P < 0.001), demonstrating non-inferiority of DRV/r qd as compared with LPV/r. The estimated difference in response for superiority of DRV/r over LPV/r was 5.5% (95% CI, -0.3-11; P = 0.062). The median change from baseline in CD4 cell count (non-completer = failure) at week 48 was similar between the groups: 137 and 141 cells/μl for DRV/r and LPV/r, respectively.
Response (HIV-1 RNA < 50 copies/ml) at week 48 was analyzed by baseline stratification factors (Fig. 2b, c). Response rates were similar for DRV/r patients irrespective of baseline HIV-1 RNA. However, in patients with high-baseline HIV-1 RNA (≥100 000 copies/ml), LPV/r response rates were lower, resulting in a statistically significantly higher response rate with DRV/r than with LPV/r (P < 0.05). Although response rates were similar across baseline CD4 cell count strata, higher numeric values were observed in the DRV/r arm compared with LPV/r, particularly in patients with fewer than 200 CD4 cells/μl (Fig. 2c, not statistically significant).
On the basis of the M-MASRI questionnaire, response rates were comparable for DRV/r patients irrespective of self-assessed adherence, whereas they differed for LPV/r.
In non-adherent patients (≦95% adherence), a numerically lower response (<50 copies/ml) at week 48 was seen with LPV/r (77%) as compared with DRV/r (90%) patients (not statistically significant). Response rates in adherent patients were similar in DRV/r and LPV/r patients (94% and 92%, respectively).
Resistance
Virologic failure (HIV-1 RNA > 50 copies/ml) at any time before the cutoff date was observed in 34 (10%) and 49 (14%) patients in the DRV/r and LPV/r arms, respectively. Baseline and endpoint (last available timepoint during treatment) genotypes were available for 10 DRV/r and 18 LPV/r virologic failures. Endpoint genotypes were not determined for the other virologic failures as the HIV-1 RNA was less than 1000 copies/ml. For one DRV/r virologic failure with HIV-1 RNA more than 1000 copies/ml, the genotype was not available at the time of the database lock. In DRV/r virologic failures, no patients developed an International AIDS Society (IAS-USA) protease inhibitor resistance-associated mutation (RAM), while one patient developed an IAS-USA nucleoside reverse transcriptase inhibitor (NRTI) RAM (M184I/V). In the LPV/r virologic failures, one patient developed two additional IAS-USA protease inhibitor RAMs (A71T and V77I) and two patients developed an IAS-USA NRTI RAM (both M184V).
Safety
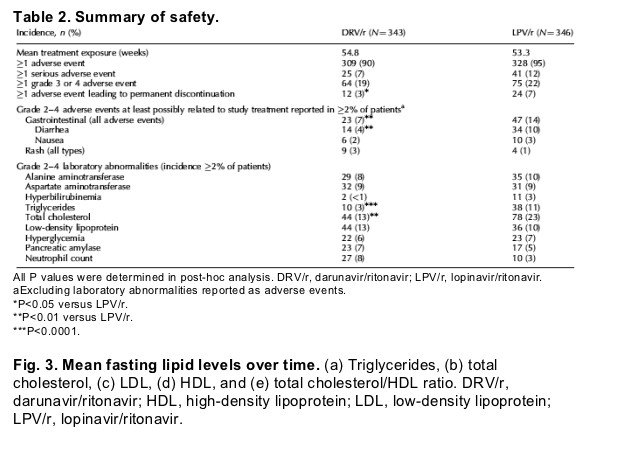
In this study, mean study drug exposures to DRV/r and LPV/r were comparable (Table 2). Most adverse events were grade 1 or 2, and discontinuations due to adverse events were infrequent; more LPV/r than DRV/r patients discontinued due to an adverse event (LPV/r 7% and DRV/r 3%; post-hoc P < 0.05). The most common adverse events (regardless of severity and causality) were diarrhea, nausea, headache, upper respiratory tract infection, nasopharyngitis, abdominal pain, vomiting, and cough. Serious adverse events (SAE) were reported in 7% of DRV/r and 12% of LPV/r patients (Table 2). Two DRV/r (<1%) and six LPV/r (2%) patients discontinued due to an SAE, which for four patients (one DRV/r and three LPV/r) was considered possibly or very likely to be treatment related. Four patients (one DRV/r and three LPV/r) died during the treatment period; none of these deaths was considered to be treatment related.
The incidence of grade 2-4 gastrointestinal-related adverse events at least possibly related to study treatment was higher for LPV/r than for DRV/r (Table 2; P < 0.01); diarrhea occurred more frequently with LPV/r than with DRV/r (P < 0.01). The overall incidence of rash-related adverse events (any grade, regardless of causality) was similar in the DRV/r (15%) and LPV/r (13%) treatment groups, and led to permanent discontinuation in less than 1% of patients for both arms. Grade 2-4 rash adverse events (all types) are shown in Table 2. There was one case of Stevens-Johnson syndrome (DRV/r arm); although other factors may have contributed to this event, a relationship with DRV/r could not be excluded. There were no serious renal adverse events reported during the study, and no patients discontinued due to renal adverse events.
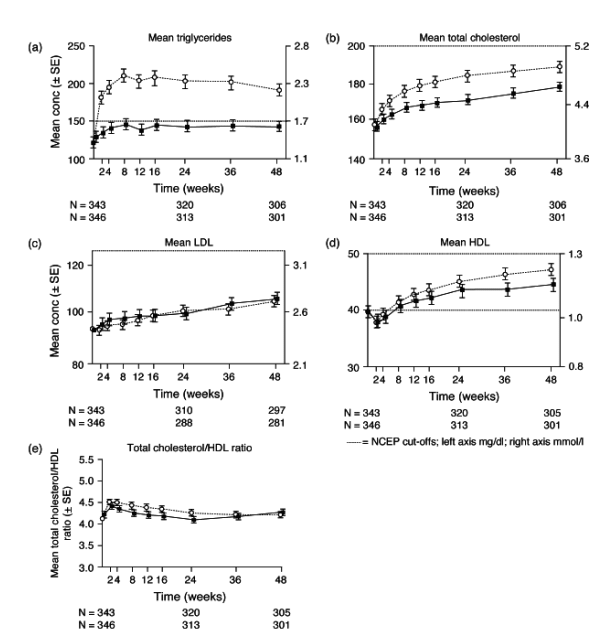
The overall incidence of laboratory abnormalities was comparable for the DRV/r and LPV/r treatment groups (Table 2). Most laboratory abnormalities were grade 1 or 2. For all lipid-related parameters in both treatment groups, small increases were seen at week 48 compared to baseline (Fig. 3). Mean increase in triglycerides and total cholesterol were more pronounced with LPV/r than with DRV/r. Changes in low-density lipoprotein (LDL) and high-density lipoprotein (HDL) cholesterol and the total cholesterol/HDL ratio were similar for both treatment groups. Grade 2-4 elevations in triglycerides and cholesterol were observed less frequently in the DRV/r (3 and 13%) than in the LPV/r group (11 and 23%; Table 2). As tenofovir was included in the background regimen, changes in calculated creatinine clearance were monitored; these changes were small and comparable between week 48 and baseline, and glomerular filtration rates were not affected.
Anthropometrics
A small weight gain was seen in patients in both treatment groups; however, this was not associated with changes in overall body shape. Although minor increases were observed in the majority of anthropometric measurements at week 48, waist-to-hip ratios were comparable to baseline for DRV/r and LPV/r. These changes are likely to be associated with restoration of health in the patients studied.
Pharmacokinetics
In all DRV/r patients evaluated (n = 335), DRV plasma concentrations exceeded the in-vitro protein-binding corrected median effective concentration required to induce 50% response (EC50) of 55 ng/ml (wild-type) [9].
Discussion
Once-daily DRV/r 800/100 mg together with a fixed NRTI background regimen was a highly effective and tolerable therapy for treatment-naive, HIV-1-infected patients. Furthermore, once-daily DRV/r was proven to be non-inferior to LPV/r (HIV-1 RNA < 50 copies/ml). All DRV/r patients achieved DRV plasma concentrations that exceeded the protein-binding corrected EC50 (wild-type) [9], suggesting that DRV/r 800/100 mg qd achieves the intended exposure in a wide range of treatment-naive patients infected with wild-type virus.
At week 48, 84% of patients reached undetectable HIV-1 RNA, providing evidence of the potency of DRV/r in this patient population and representing one of the highest response rates observed in treatment-naive HIV patients in trials to date [10,11]. The efficacy of DRV/r qd was not compromised in the subgroups of patients that are typically more prone to virologic failure (high HIV-1 RNA, low CD4 cell count or non-adherent patients): patients with high pretreatment HIV-1 RNA were more likely to respond to DRV/r qd than LPV/r (qd or bid), demonstrating the robustness of this regimen.
In this study, DRV/r 800/100 mg qd was generally safe and well tolerated, with few treatment discontinuations, and the type and incidence of adverse events in the DRV/r qd group are consistent with those reported in trials of treatment-experienced patients [1-5] that showed a favorable gastrointestinal tolerance, specifically in terms of a lower incidence of diarrhea. In this study, gastrointestinal adverse events were more frequent in LPV/r than DRV/r patients. Adverse events were not compared by LPV/r formulation due to the low number of patients initiating with tablets (n = 6). The incidence of rash was similar in the two treatment groups, in line with previous studies [1-5] and rarely led to discontinuation. There were no serious renal adverse events or discontinuations due to renal adverse events in this study, despite the fact that DRV/r and LPV/r have been reported to increase serum levels of tenofovir. Triglyceride increases were more frequent with LPV/r than DRV/r, which may be related to the higher daily dose of ritonavir with LPV/r.
It should be noted that some flexibility in dosing and formulation of LPV/r was permitted within the study protocol, based on local regulatory approval and the preferences of investigators and patients. However, given that bioequivalence between the capsule and tablet formulations of LPV/r has been demonstrated [12], and also that a recent large, randomized, controlled trial established non-inferiority of qd as compared with bid dosing of LPV/r with no differences in efficacy or safety in treatment-naive patients [13], this is not expected to influence our findings.
In conclusion, patients receiving once-daily DRV/r achieved high durable virologic response rates, (which were comparable in patients with less favorable baseline characteristics or suboptimal adherence), had a low rate of discontinuation due to virologic failure or adverse events or both, did not develop protease inhibitor resistance upon failure, and had suitable drug exposure. These benefits, coupled with the favorable safety and pharmacokinetic profile of DRV/r, suggest that DRV/r 800/100 mg qd has the potential to become a first-line, once-daily treatment option for treatment-naive patients.
|
|
| |
| |
|
|
|